
Opinion statement
Cardiovascular disease is a leading cause of morbidity and mortality in the USA and around the world. While we are now able to achieve significant low-density lipoprotein cholesterol (LDL-C) lowering with current therapies, many patients remain at risk for cardiovascular disease (CVD). Elevated lipoprotein(a) [Lp(a)] has been shown to be an independent risk factor for CVD and accounts for some of the residual CVD risk after LDL-C lowering in several large clinical trials. Moreover, there is now strong evidence supporting the causal relationship between Lp(a) and aortic stenosis as well as peripheral arterial disease. Despite the growing interest in this lipoprotein, the current therapeutic options for Lp(a) reduction are limited. Our general approach in patients with elevated Lp(a) levels is to aggressively manage other modifiable cardiovascular risk factors including lifestyle modification, consideration of aspirin therapy, and LDL-C lowering. Unfortunately, there are conflicting reports on how effective this strategy is at reducing the risk for cardiovascular events attributed to elevated Lp(a). As a result, targeted Lp(a)-lowering strategies are needed. Lp(a) therapeutics is an active area of research with several promising classes of pharmacotherapies under investigation to address this causal biomarker.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease (CVD) is the leading cause of death in the USA [1]. Low-density lipoprotein cholesterol (LDL-C) is a well-established, major risk factor for CVD and large, randomized, trials with HMG Co-A reductase inhibitors (statins) have demonstrated a reduction in major coronary events and coronary death with LDL-C lowering [2]. However, despite high intensity statin therapy, patients continue to have residual risk, with some studies showing major cardiovascular event rates of 8 to 9% over 3 to 5 years [3, 4]. While many factors undoubtedly contribute to this, there is growing evidence that lipoprotein (a) [Lp(a)] could explain a proportion of this residual risk.
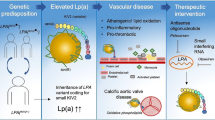
Lp(a) is a circulating lipoprotein composed of the liver-derived apolipoprotein (a) [apo(a)] covalently bound to apolipoprotein B-100 [apoB] of an LDL particle [5•]. The LPA gene is located on chromosome 6 next to the plasminogen gene (PLG), from which it arose after a gene duplication event [6]. The LPA gene codes for multiple kringle structures, protein domains that fold into large loops that are highly homologous to PLG. Unlike PLG, however, LPA kringle number is highly heterogeneous. The number of kringle IV-type 2 copies is inversely correlated to plasma Lp(a) concentration [5•]. Lp(a) is cleared primarily by the liver through an LDL-receptor-independent mechanism [7]. There is also evidence for some renal catabolism [8].
Relationship to cardiovascular disease
Over the last several decades, a number of large epidemiological studies, meta-analyses, and Mendelian randomization studies have suggested a causal link between Lp(a) concentration and myocardial infarction and ischemic stroke [9,10,11,12, 13•, 14]. The risk is higher after approximately 30 mg/dL and is thought to be independent [15]. The association is stronger at higher Lp(a) concentrations, especially above 50 mg/dL [16]. In the Copenhagen City Heart Study, subjects at the top 95th percentile for Lp(a) concentration (>120 mg/dL) had more than a threefold increased risk of developing a myocardial infarction compared with those in the lowest Lp(a) quartile [17]. Although relatively few African Americans were studied in many past investigations, a study by Virani and colleagues in 2012 showed an elevated risk of CVD in African American individuals with high Lp(a) concentrations [18]. Moreover, an elevated Lp(a) concentration has been shown to be an independent predictor of CVD in familial hypercholesterolemia (FH). In the Copenhagen General Population Study, the hazard ratio for myocardial infarction in non-FH/elevated Lp(a) was 1.4; in FH/normal Lp(a) 3.2 and in FH/elevated Lp(a) was 5.3 (all relative to non-FH/normal Lp(a)) [19]. Lp(a) has been demonstrated to be higher in FH patients than their unaffected relatives, but ascertainment bias cannot be ruled out [20].
In addition to the independent association with stroke and coronary disease, Lp(a) has been associated with aortic stenosis. In 2013, Thanassoulis and colleagues showed that genetic variation in the LPA gene is strongly associated with aortic valve calcification and stenosis [21]. A subsequent Mendelian randomization study design showed that genetically elevated Lp(a) levels were associated with an increased risk of aortic valve stenosis, with levels >90 mg/dL predicting a threefold increased risk [22]. Taken together, the evidence demonstrates a causal relationship between elevated Lp(a) concentration and cardiovascular disease, including myocardial infarction, ischemic stroke, peripheral arterial disease, and aortic stenosis. Indeed, a recent study of the phenotypic impact of LPA gene variation found that one standard deviation of genetically lowered Lp(a), which translated to a 28 mg/dL reduction in Lp(a), is associated with a 29% reduction in risk for coronary heart disease, 13% reduction in risk of stroke, 31% reduction in peripheral arterial disease, and 37% reduction in risk of aortic stenosis with no association with type 2 diabetes, cancer, or 31 other disorders [23••]. Thus, pharmacologic reduction of Lp(a) may impact a range of cardiovascular disease traits.
Pathophysiology
The increased CVD risk of Lp(a) is mediated through proatherogenic and prothrombotic mechanisms [24] (Table 1). With regard to proatherogenic mechanisms, it is believed that Lp(a) may be more avidly retained within arterial walls than LDL-C [25]. Lp(a) is also a preferential carrier of proinflammatory oxidized phospholipids (OxPL), which accumulate in atherosclerotic lesions and mediate plaque destabilization [26, 27]. Lp(a) is also thought to promote atherogenesis through increasing endothelial cell permeability, promoting smooth muscle proliferation, and macrophage foam cell formation [28]. There are various mechanisms by which Lp(a) promotes thrombosis. As previously noted, apo(a) is highly homologous to plasminogen and therefore may interfere with fibrin clot lysis through impeding the conversion of plasminogen to plasmin [28]. Lp(a) has also been shown to bind and inactivate tissue factor pathway inhibitor, which is a major regulator of the tissue factor-mediated coagulation pathway [28].
Plasma Lp(a) concentrations are largely genetically determined, though the exact genetic contribution is unknown [29, 30]. In the general population, Lp(a) levels vary greatly among individuals and between ethnicities [31•]. For example, Lp(a) concentrations are higher in individuals of African descent compared with European and Asian descent [32,33,34,35,36]. It is estimated that 30% of Caucasians and 60 to 70% of African Americans have Lp(a) levels >25 mg/dL [37]. In Europeans and Asians, Lp(a) concentrations are highly skewed with a tail toward higher concentrations [35]. A recent study of over 500,000 patients referred for Lp(a) measurement had a median Lp(a) value of 17 mg/dL with an interquartile range of 7–47 mg/dL. Of those patients analyzed at a tertiary referral center, 39.5% had an Lp(a) greater than 30 mg/dL and 29.2% had a level greater than 50 mg/dL [38].
Lp(a) measurement
Importantly, measurement of Lp(a) concentration is not fully standardized and is performed using several assays. One category of assays is “isoform-independent” utilizing an antibody to a non-repeating kringle. Thus, it measures Lp(a) particle number and is reported in units of nanomoles per liter. The other category is “isoform-dependent,” representing the entire protein mass of Lp(a), reported in milligrams per deciliter. Since Lp(a) isoforms with more kringle repeats will have more mass, a mismatch develops between mass and particle number for larger isoforms. Therefore, the isoform-independent assays are the preferred measurement method given that apo(a) size will not affect the result [37]. Notably, fasting and non-fasting Lp(a) levels appear to be similar [39]. Currently, most Lp(a) guidelines report Lp(a) concentration in milligrams per deciliter with an upper limit of normal of 30 mg/dL [40]. Assays that report Lp(a) concentration in nanomoles per liter typically use an upper limit of normal of 75 nmol/L [41].
Screening
The strong association between Lp(a) and CVD has prompted some professional groups to publish guidelines regarding Lp(a) screening, but recommendations are not standardized. Both the National Lipid Association and European Atherosclerosis Society suggest that the following groups be screened for elevated Lp(a) concentrations: those with a personal or family history of premature CVD, familial hypercholesterolemia, recurrent CVD events despite statin therapy, ≥3% 10-year risk of fatal CVD by European guidelines or ≥10% 10-year risk of fatal, and/or non-fatal heart disease per US guidelines [40, 42]. The 2012 Canadian Cardiovascular Society recommends consideration of secondary testing, including Lp(a), in those with a 10 to 19% Framingham Risk [43]. The National Lipid Association does not recommend screening for Lp(a) levels in low-risk patients (less than 5% 10-year Framingham Risk) [40].
Treatment
Diet and lifestyle
Healthy dietary interventions have been shown to reduce atherogenic lipids and lipoproteins with diets rich in mono- and poly-unsaturated fats or protein showing the greatest benefits. Unfortunately, the summation of currently available evidence shows that dietary interventions have no appreciable effect on Lp(a) levels [37, 44, 45].
Similarly, current data from cross-sectional and interventional studies show that moderate exercise has little effect on Lp(a) concentrations [46]. Lp(a) concentrations may actually be elevated in athletes who exercise intensely on a daily basis [46]. In another small study, there was a 13% Lp(a) reduction with exercise in patients with baseline Lp(a) concentrations >30 mg/dL [47]. There was no significant Lp(a) reduction in the whole group analysis.
Although lifestyle factors may not directly impact Lp(a) levels, ideal lifestyle does, to a degree, mitigate genetic risk and should always be the foundation of cardiovascular preventive care [48].
Medical therapy not directed at Lp(a)
Before discussing specific pharmacotherapies aimed at targeting Lp(a) directly, it is important to outline the approach to overall cardiovascular risk management in those with elevated Lp(a), namely, LDL-C reduction with statins and antiplatelet therapy with aspirin.
Several studies have shown that in patients with elevated Lp(a), CVD risk is greater in those with high LDL-C compared to those with low LDL-C [49,50,51]. A study by Nicholls and colleagues found no enhanced risk for CVD in patients with both elevated Lp(a) and LDL-C < 70 mg/dL [51]. These results support the general consensus that intensified lipid management should be initiated in those with elevated Lp(a) despite the observations that statins have either no effect or increase Lp(a) concentrations [52]. However, given current limited evidence, among patients with primary Lp(a) elevations without other indications for statin therapy per the 2013 American College of Cardiology/American Heart Association guidelines, the benefit of statin therapy for CVD risk reduction is unclear [41, 53].
Given the prothrombotic nature of elevated Lp(a), it has been postulated the aspirin therapy would help reduce CVD risk. A relatively small study conducted in 2002 by Akaike and colleagues showed an 88.3% decrease in Lp(a) levels in patients with baseline Lp(a) >30 mg/dL who took aspirin therapy. There was no statistically significant decrease in patients with Lp(a) concentrations less than 30 mg/dL [54]. No subsequent studies have been able to reproduce this effect. However, Chasman and colleagues showed in the Women’s Heart Study that women who carried an LPA locus genetic variant (3.7% of the cohort and associated with fivefold higher median Lp(a) levels) and took aspirin had a 56% risk reduction of major cardiovascular events [55]. By comparison, those women without the risk allele who took aspirin had no significant risk reduction. Though hopeful, there is an overall lack of evidence for improved outcomes with aspirin therapy in those with elevated Lp(a) and no professional guidelines base their recommendation for aspirin therapy on Lp(a) level.
Pharmacologic treatment
While it is currently unknown whether lowering Lp(a) will translate into decreased cardiovascular events, the robust evidence causally linking elevated Lp(a) levels to cardiovascular disease has led to the development of pharmacologic approaches to lower Lp(a). Below, we review the available and investigational therapies that target, or indirectly lower, Lp(a). They are also summarized in Table 2.
Niacin
At therapeutic doses, niacin lowers LDL-C, raises high-density lipoprotein cholesterol (HDL-C), lowers triglycerides, and also lowers Lp(a) [56]. Recent clinical trials of randomization to niacin in subjects already receiving standard statin therapy, however, have failed to show benefit [57, 58].
Nonetheless, based on a meta-analysis that recently showed positive effects of niacin alone or in combination with other lipid-lowering drugs on all cardiovascular events and on atherosclerosis evolution, the European Atherosclerosis Society recommends the use of niacin at a dose of 1–3 g per day in high-risk patients after suitable LDL-cholesterol reduction to achieve an Lp(a) concentration less than 50 mg/dL [42, 59]. These treatment aims are considered secondary to the main goal of reducing LDL-C and total cholesterol levels [42].
Hormone replacement therapy
Observational studies in the early 1990s found that Lp(a) levels increase after menopause, which raised interest in the possible effects of hormone replacement therapy (HRT) on Lp(a) levels [60]. Indeed, both estrogens and progestins have been shown to reduce Lp(a) [61,62,63]. Most of these studies were conducted in small cohorts with short follow-up periods. Importantly, given that several more recent studies have shown that HRT may increase CVD risk and does not uniformly confer cardiac protection, the use of HRT for Lp(a) reduction or prevention of CVD is not recommended [64].
Mipomersen
Mipomersen is an apoB antisense oligonucleotide that is FDA approved as an adjunct for lowering LDL-C, apoB, total cholesterol, and non-HDL-C in patients with homozygous FH [65]. Antisense oligonucleotides are synthetic analogs of nucleic acids designed to bind specific messenger RNA, suppressing the synthesis of its protein product. Mipomersen targets apoB and impairs the formation of very-low-density lipoproteins, consequently lowering LDL-C by approximately 25% [65]. A recent analysis of data from four phase III trials found that mipomersen also decreased Lp(a) by 26.4% [66]. Due to concerns about hepatotoxicity, mipomersen can only be prescribed by registered specialists. Currently, as previously mentioned, mipomersen is only FDA approved for the treatment of homozygous FH.
Lomitapide
Lomitapide is a microsomal triglyceride transfer protein (MTP) inhibitor with FDA approval as adjunct lipid-lowering therapy to lower LDL-C, total cholesterol, apoB, and non-HDL-C in patients with homozygous FH [67]. MTP is an intracellular protein responsible for the transfer of lipid molecules onto apoB. This process is important for the assembly of chylomicrons and very-low-density lipoproteins. Similarly, patients with abetalipoproteinemia have loss-of-function mutations in the microsomal triglyceride transfer protein gene resulting in extremely low plasma cholesterol and triglycerides as well as the absence of chylomicrons, very-low-density lipoproteins, and LDL-C [68]. This discovery led to the concept of small-molecule inhibitors of MTP with the goal of reducing plasma cholesterol levels.
In an open-label, phase III study investigating the safety and efficacy of lomitapide in patients with homozygous FH, the drug showed a 15% reduction in Lp(a) at 26 weeks, which persisted to 56 weeks. However, by 78 weeks, there was no statistically significant difference in Lp(a) concentration from baseline [69]. Like mipomersen, significant hepatotoxicity has been seen with lomitapide, and therefore, the drug must also be prescribed by a registered specialist. Moreover, the long-term clinical outcomes data for lomitapide are not yet available. Currently, as previously mentioned, lomitapide is only FDA approved for the treatment of homozygous FH.
Proprotein convertase subtilisin-kexin type 9 inhibitors
In 2015, the FDA approved alirocumab and evolocumab for clinical use. These first-in-class medications are fully humanized monoclonal antibodies that inhibit proprotein convertase subtilisin-kexin type 9 (PCSK9). PCSK9 was discovered when investigators found gain-of-function mutations in PCSK9 in families with autosomal dominant familial hypercholesterolemia [70]. Subsequently, it was shown that loss-of-function mutations in PCSK9 lead to low LDL-C and reduced risk for coronary artery disease [71]. Genetic and pharmacologic reduction results in decreased LDL-receptor degradation, increased recirculation of the receptor to the surface of hepatocytes, and ultimately lowering of serum LDL-C. These drugs have been shown to reduce LDL-C by up to approximately 70% [72, 73]. The current FDA indications for PCSK9 inhibitors include inadequate LDL-cholesterol lowering despite diet and maximally tolerated statins in those with known CVD, heterozygous familial hypercholesterolemia, or homozygous FH (evolocumab only).
In addition to lowering LDL-C, PCSK9 inhibitors have also been shown to lower Lp(a) concentrations. An analysis evaluated the effect of alirocumab on Lp(a) levels in pooled data from three double-blind, randomized, placebo-controlled, phase II studies of 8 or 12 weeks duration in patients with hypercholesterolemia and background lipid-lowering therapy [74•]. Alirocumab resulted in a 30.3% reduction in Lp(a) concentration from baseline in this study, which was significant compared with the 0.3% reduction observed in the placebo group [74•].
Similar Lp(a) lowering has been shown with evolocumab. A pooled analysis of data from 1359 patients in four phase II trials assessing the effects of evolocumab over a 12-week period found up to a 29.5% reduction in Lp(a) with no plateau effect [75•].
The mechanism of action by which PCSK9 inhibitors lower Lp(a) is unclear. In vitro cultured hepatocyte studies demonstrated reduced production (hypothesized reduced association of apo(a) with apoB at the hepatocyte cell surface) rather than increased clearance with alirocumab [76]. However, stable isotope studies in normal volunteers demonstrated increased clearance, not production, with alirocumab [77].
While there appears to be reproducible evidence of Lp(a) reductions with PCSK9 inhibitors, it remains to be seen whether these drugs will be used for this indication.
Other treatments
Apheresis
Apheresis is a process that removes all apoB-containing lipoproteins from the blood and remains the most effective current treatment modality to lower Lp(a), with acute reductions of up 75% [78]. In a longitudinal, multicenter, cohort study of 120 patients with CVD and Lp(a) levels in the 95th percentile or greater, maximally tolerated medical therapy plus apheresis resulted in a median Lp(a) reduction of 73%, which was a statistically significant reduction compared with medical therapy alone [79]. Moreover, the study showed that combination therapy with apheresis reduced major adverse cardiac events by 88% over a 10-year follow-up period [79]. Similar results were reported in a recent prospective observational trial of 170 patients, where one apheresis treatment reduced Lp(a) by an average of 68.1%, and treatment over the course of 5 years significantly reduced annual cardiovascular event rates [80].
One significant disadvantage of apheresis is that lipoprotein levels tend to rebound to baseline within 2 weeks of treatment, necessitating frequent sessions. Moreover, while apheresis has been shown to reduce major adverse cardiac events, it is not possible to ascribe this benefit to Lp(a) reduction alone, as the treatment lowers all lipoprotein levels. Finally, the significant cost and limited access to apheresis centers pose significant challenges to the more widespread use of this technology.
Emerging therapies
Cholesteryl ester transfer protein inhibitors
Cholesteryl ester transfer protein (CETP) is a plasma protein that promotes the transfer of cholesteryl esters from HDL-C to other lipoproteins. The inhibition of this protein raises HDL-C and decreases LDL-C and Lp(a). The first large clinical trial with the CETP inhibitor torcetrapib was terminated due to an increase in cardiovascular events and all-cause death in the treatment arm [81]. A phase III study of the CETP inhibitor dalcetrapib showed increased HDL-C levels, but no reduced risk of recurrent cardiovascular events in the treatment group [4]. Recently, a randomized trial studying the CETP inhibitor evacetrapib was terminated after the drug was not shown to be superior to placebo in reducing cardiovascular outcomes despite favorable effects on HDL-C and LDL-C [82].
In the phase III efficacy and safety study with CETP inhibitor anacetrapib, the drug showed a sustained lowering of Lp(a) by 38.8% from baseline levels [83]. The Randomized Evaluation of the Effects of Anacetrapib through Lipid-modification (REVEAL) trial is a phase III study of 30,000 patients designed to determine whether lipid modification with this drug reduces major cardiovascular outcomes in patients with CVD on background statin therapy. The study is estimated to be completed in January 2017 [84].
The negative results for torcetrapib, dalcetrapib, and evacetrapib suggest that CETP inhibition may not be an effective strategy to mitigate residual cardiovascular risk. The REVEAL trial is ongoing and may help guide whether the pursuit of CETP inhibitors will continue.
Antisense oligonucleotides targeting apolipoprotein(a)
There have been significant efforts to develop Lp(a)-specific pharmacotherapies. The first such class of agents are antisense oligonucleotides targeting hepatic apolipoprotein(a) mRNA to lower Lp(a) plasma concentrations. In a pre-clinical mouse model study of ASO 144367, the molecule resulted in significant reductions in both Lp(a) and apo(a). Specifically, Lp(a) was reduced by 24.8%, and apo(a) levels decreased by 19.2 to 86%, with the more prominent effect seen in mice expressing apo(a) with more KIV-2 repeats [85].
In a recent randomized, double-bind, placebo-controlled, phase I study of 47 healthy adults with baseline Lp(a) concentrations of 25 nmol/L or greater, six doses of the ASO molecule IONIS-APO(a)Rx (previously called ISIS-APO(a)Rx) resulted in dose-dependent mean percentage decreases in plasma Lp(a) concentration of 39.6 to 77.8%. Mild injection site reactions were the most common adverse events [86].
Based on the promising findings in this phase I study, two follow-up trials were conducted [87•]. The first was a phase II trial of IONIS-APO(a)Rx. A total of 64 patients were randomized to escalating-dose subcutaneous IONIS-APO(a)Rx once for 4 weeks each or placebo. At 85 or 99 days, based on the per-protocol population, participants assigned to IONIS-APO(a)Rx had mean Lp(a) reductions of 66.8 or 71% (the former in those with baseline Lp(a) concentrations from 125 to 437 nmol/L and the latter in those with concentrations ≥438 nmol/L), which were significant compared with the pooled placebo.
This trial was followed by a phase I/IIa first-in-man trial of IONIS-APO(a)-LRx, a ligand-conjugated antisense oligonucleotide designed to be highly and selectively taken up by hepatocytes. A total of 58 healthy volunteers with Lp(a) ≥ 75 nmol/L were randomly assigned to IONIS-APO(a)-LRx subcutaneously in single or multiple dose subgroups or placebo. Significant dose-dependent reductions in Lp(a) concentrations were noted in all single-dose IONIS-APO(a)-LRx groups at day 30. In the multidose groups, IONIS-APO(a)-LRx resulted in mean reductions in Lp(a) between 66% and 92%, based on dose. In both trials, LDL-C and apoB were also reduced. Both antisense oligonucleotides were deemed safe.
Overall, antisense-mediated targeting of apo(a) mRNA is a promising new frontier in Lp(a) pharmacotherapy. To date, it is the most potent and specific therapy to lower Lp(a). Larger and long-term trials will be necessary to determine whether such therapies could mitigate Lp(a)-mediated cardiovascular risk.
Interleukin-6 receptor antagonists
Several studies have revealed that Lp(a) induction can be caused by chronic inflammation [87•]. This is supported by the observation that several chronic inflammatory diseases are associated with elevated Lp(a) serum levels [88]. In a study of 11 patients with rheumatoid arthritis treated with tocilizumab, a monoclonal antibody against interleukin-6 (IL-6), there was a statistically significant decrease in Lp(a) levels by up to 50% [89]. Similar results were published in an independent cohort [90]. Interestingly, subsequent studies have shown that adalimumab, the tumor necrosis factor-α-inhibitor, has no such effect on Lp(a) levels [91]. The interaction between Lp(a) and chronic inflammation is an emerging construct with potential for pharmacologic interventions in the future, but further investigation is required.
References and Recommended Reading
Recently published papers of particular interest have been denoted as: • Of importance •• Of outstanding importance
CDC, NCHS. Underlying Cause of Death 1999–2013 on CDC WONDER Online Database, released 2015. Data are from the Multiple Cause of Death Files, 1999–2013, as compiled from data provided by the 57 vital statistics jurisdictions through the Vital Statistics Cooperative Program. Accessed Nov. 5, 2016.
Cholesterol Treatment Trialists (CTT) collaboration. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385:1397–405.
Mora S, Wenger NK, DeMicco DA, Breazna A, Boekholdt SM, Arsenault BJ, Deedwania P, Kastelein JJP, Waters DD. Determinants of residual risk in secondary prevention patients treated with high- versus low-dose statin therapy: the treating to new targets (TNT) study. Circulation. 2012;125:1979–87.
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJV, Mundl H, Nicholls SJ, Shah PK, Tardif J-C, Wright for the dal-OUTCOMES Investigators. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med 2012;367:2089–2099.
• Lamon-Fava S, Diffenderfer MR, Marcovina SM. Lipoprotein(a) metabolism. Curr Opin Lipidol. 2014;25:189–93. Excellent review of lipoprotein(a) metabolism
Lawn RM, Schwartz K, Patthy L. Convergent evolution of apolipoprotein(a) in primates and hedgehog. Proc Natl Acad Sci. 1997;94:11992–7.
Cain WJ, Millar JS, Himebauch AS, et al. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a]. J Lipid Res. 2005;46:2681–91.
Schmidt K, Noureen A, Kronenberg F, et al. Structure, function and genetics of lipoprotein(a). J Lipid Res. 2016;56:139–59.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, et al. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–9.
Erqou S, Thompson A, Di Angelantonio E, et al. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol. 2010;55:2160–7.
Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56:946–55.
Kiechl S, Willeit J, Mayr M, et al. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Atherioscler Thromb Vasc Biol. 2007;27:1788–95.
• Beheshtian A, Shitole SG, Segal AZ, et al. Lipoprotein(a) level, apolipoprotein size, and risk of unexplained ischemic stroke in young and middle-aged adults. Atherosclerosis. 2016;253:47–53. Cryptogenic stroke in younger adults is a clinical conundrum under active investigation. This meta-analysis discusses the potential role of Lp(a)
Nave AH, Lange KS, Leonards CO, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015;242:496–503.
Erqou S, Kaptoge S, Perry PL, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–23.
Kolski B, Tsimikas S. Emerging therapeutic agents to lower lipoprotein (a) levels. Curr Opin Lipidol. 2012;23:560–8.
Kamstrup PR, Benn M, Tybjaerg-Hansen A, et al. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117:176–84.
Virani SS, Brautbar S, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125:241–9.
Langsted A, Kamstrup PR, Benn M, et al. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol. 2016;4:577–87.
Alonso R, Andres E, Mata N, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63:1982–9.
Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcific and aortic stenosis. N Engl J Med. 2013;368:503–12.
Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63:470–7.
•• Emdin CA, Khera AV, Natarajan P, et al. Phenotypic characterization of genetically lowered human lipoprotein(a) levels. J Am Coll Cardiol. 2016;68:2761–72. The potential impact of lowering Lp(a), as determined from LPA gene variants, on cardiovascular disease is discussed
Boffa MB, Koschinsky ML. Update on lipoprotein(a) as a cardiovascular risk factor and mediator. Curr Atheroscler Rep. 2013;15:360.
Nielsen LB. Atherogenecity of lipoprotein(a) and oxidized low density lipoprotein: insight from in vivo studies of arterial wall influx, degradation and efflux. Atherosclerosis. 1999;143:229–43.
Tsimikas S, Brilakis ES, Miller ER, et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353:46–57.
Bergmark C, Dewan A, Orsoni A, et al. A novel function of lipoprotein [a] as preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–9.
Marcovina SM, Koschinsky ML. Evaluation of lipoprotein(a) as a prothrombotic factor: progress from bench to bedside. Curr Opin Lipidol. 2003;14:361–6.
Lanktree MB, Anand SS, Yusuf S, on behalf of the SHARE Investigators. Comprehensive analysis of genomic variation in the LPA locus and its relationship to plasma lipoprotein (a) in south Asians, Chinese and European Caucasians. Circ Cardiovasc Genet. 2010;3:39–46.
Rao F, Schork AJ, Maihofer AX, et al. Heritability of biomarkers of oxidized lipoproteins: twin pair study. Arterioscler Thromb Vasc Biol. 2015;35:1704–11.
• Nordestgaard BG, Langsted A. Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res. 2016;57:1953–75. Excellent review of Lp(a) genetics, epidemiology, pathophysiology, and association with cardiovascular disease and aortic stenosis
Enkhmaa B, Anuurad E, Berglund L. Lipoprotein (a): impact by ethnicity and environmental and medical conditions. J Lipid Res. 2016;57:1111–25.
Sandholzer C, Hallman DM, Saha N, et al. Effects of the apolipoprotein(a) size and polymorphism on lipoprotein(a) concentration in 7 ethnic groups. Hum Genet. 1991;86:607–14.
Kraft HG, Lingenhel A, Pang RW, et al. Frequency distributions of apoliporpotein(a) kringle IV repeat alleles and their effects on lipoprotein(a) levels in Caucasian, Asian, and African populations: the distribution of null alleles is non-random. Eur J Hum Genet. 1996;4:74–87.
Gaw A, Boerwinkle E, Cohen JC, et al. Comparative analysis of the apo(a) gene, apo(a) glycoprotein, and plasma concentrations of Lp(a) in three ethnic groups. Evidence for no common “null” allele at the apo(a) locus. J Clin Invest. 1994;93:2526–34.
Marcovina SM, Albers JJ, Wijsman E, et al. Differences in Lp[a] concentrations and apo(a) polymorphs between black and white Americans. J Lipid Res. 1996;37:2569–85.
Tsimikas S, Hall JL. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease. A rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol. 2012;60:716–21.
Varvel S, McConnell JP, Tsimikas S. Prevalence of elevated Lp(a) mass levels and patient thresholds in 532,359 patients in the United States. Arterioscler Thromb Vasc Biol. 2016;36:2239–45.
Langsted A, Kamstrup PR, Nordestgaard BG. Lipoprotein(a): fasting and nonfasting levels, inflammation, and cardiovascular risk. Atherosclerosis. 2014;234:95–101.
Davidson MH, Ballantyne CM, Jacobson TA, et al. Clinical utility of inflammatory markers and advanced lipoprotein testing: advice from an expert panel of lipid specialists. J Clin Lipidol. 2011;5:338–67.
Mann LC, Kelly E, Duffy D. Targeting lipoprotein (a): an evolving therapeutic landscape. Curr Atheroscler Rep. 2015;17:25.
Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–53.
Anderson TJ, Gregoire J, Hegele RA, et al. 2012 update of the Canadian Cardiovascular Society guidelines for the diagnosis and treatment of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol. 2013;29:151–67.
Haring B, Wyler von Ballmoos M, Appel LJ, et al. Healthy dietary interventions and lipoprotein (a) plasma levels: results from the Omni Heart Trial. PLoS One. 2014;9(12):1–12.
Hirowatari Y, Manita D, Kamachi, et al. Effect of dietary modification by calorie restriction on cholesterol levels in lipoprotein(a) and other lipoprotein classes. Ann Clin Biochem. 2016.
Mackinnon LT, Hubinger L, Lepre F. Effects of physical activity and diet on lipoprotein(a). Med Sci Sports Exerc. 1997;29(11):1429–36.
Rigla M, Sánchez-Quesada JL, Ordóñez-Llamos J, et al. Effect of physical exercise on lipoprotein(a) and low-density lipoprotein modifications in type 1 and type 2 diabetic patients. Metabolism. 2000;49(5):640–7.
Khera AV, Emdin CA, Drake I, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016; doi:10.1056/NEJMoa1605086.
Luc G, Bard JM, Arveiler D, et al. Lipoprotein (a) as a predictor of coronary heart disease: the PRIME Study. Atherosclerosis. 2002;163:377–84.
Maher VM, Brown BG, Marcovina SM, et al. Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein(a). JAMA. 1995;274:1771–4.
Nicholls SJ, Tang WHW, Scoffone H, et al. Lipoprotein(a) levels and long-term cardiovascular risk in the contemporary era of statin therapy. J Lipid Res. 2010;51:3055–61.
Taleb A, Witztum JL, Tsimikas S. Oxidized phospholipids on apolipoprotein B-100 (OxPL/apoB) containing lipoproteins: a biomarker predicting cardiovascular disease and cardiovascular events. Biomark Med. 2011;5:673–94.
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S1–45.
Akaike M, Azuma H, Kagawa A, et al. Effect of aspirin treatment on serum concentrations of lipoprotein(a) in patients with atherosclerotic diseases. Clin Chem. 2002;48:1454–9.
Chasman DI, Shiffman D, Zee RY, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009;203:371–6.
Carlson LA, Hamsten A, Asplund A. Pronounced lowering of serum levels of lipoprotein Lp(a) in hyperlipidemic subjects treated with nicotinic acid. J Intern Med. 1989;226:271–6.
Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12.
The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
Bruckert E, Labreuche J, Amarenco P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis. 2010;210:353–61.
Meilhahn EN, Kuller LH, Matthews KA, et al. Lp(a) concentrations among pre- and postmenopausal women over time: the Healthy Women Study. Circulation. 1991;84(suppl II:II-2170.Abstract.
Sacks FM, McPherson R, Walsh BW. Effect of postmenopausal estrogen replacement on plasma Lp(a) lipoprotein concentrations. Arch Intern Med. 1994;154:1106–10.
Soma M, Fumagalli R, Paoletti R, et al. Plasma Lp(a) concentration after oetrogen and progestogen in postmenopausal women. Lancet. 1991;337:612.
Kim CJ, Jang C, Cho DH, et al. Effects of hormone replacement therapy on lipoprotein(a) and lipids in postmenopausal women. Arterioscler Thromb. 1994;14:275–81.
Manson JE, Hsia J, Johnson KC, et al. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med. 2003;349:523–34.
Kynamro™(mipomersen sodium) injection [package insert]. Cambridge, MA. Genzyme. Revised January 2015.
Santos RD, Raal FJ, Catapano AL, et al. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: results of 4 phase III trials. Arterioscler Thromb Vasc Biol. 2015;35:689–99.
Juxtapid (lomitapide) capsules [package insert]. Cambridge, MA. Aegerion Pharmaceuticals. Revised August, 2014.
Rader DJ, Brewer HB. Abetalipoproteinemia: new insights into lipoprotein assembly and vitamin E metabolism from a rare genetic disease. JAMA. 1993;270:865–9.
Cuchel M, Meagher EA, du Toit TH, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6.
Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6.
Cohen JC, Boerwinkle E, Mosley TH, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2013;354:1264–72.
Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99.
Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–9.
• Gaudet D, Kereiakes DJ, McKenney JM, et al. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol. 2014;114:711–5. An analysis of alirocumab’s effect on Lp(a) levels in pooled data from three phase II studies. The authors report a 30.3% reduction in Lp(a) concentration from baseline
• Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cariol. 2014;63:1278–88. An analysis of evolocumab’s effect on Lp(a) levels in pooled data from four phase II studies. The authors report a 29.5% reduction in Lp(a) concentration from baseline (for 140mg, dosed every 2 weeks)
Villard EF, Thedrez A, Blankenstein J, et al. PCSK9 modulates the secretion but not the cellular uptake of lipoprotein(a) ex vivo: an effect blunted by alirocumab. JACC Basic Trans Science. 2016;1:419–27.
Reyes-Soffer G, Pavlyha M, Ngai C, et al. Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation. 2017;135:352–62.
Arai K, Orsoni A, Mallat Z, et al. Acute impact of apheresis on oxidized phospholipids in patients with familial hypercholesterolemia. J Lipid Res. 2012;53:1670–8.
Jaeger BR, Richter Y, Nagel D, et al. Longitudinal cohort study on the effectiveness of lipid apheresis treatment to reduce high lipoprotein(a) levels and prevent major adverse coronary events. Nat Clin Pract Cardiovasc Med. 2009;6(3):299–39.
Roeseler E, Julius U, Spitthoever R, et al. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36:2019–27.
Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22.
American College of Cardiology. Latest in Cardiology. Assessment of Clinical Effects of Cholesteryl Ester Transfer Protein Inhibition With Evacetrapib in Patients at a High Risk for Vascular Outcomes – ACCELERATE. 2016 Aug 29. Available at: http://www.acc.org/latest-in-cardiology/clinical-trials/2016/03/29/22/34/accelerate#references-for-article
Cannon CP, Shah S, Dansky HM, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–15.
REVEAL: randomized evaluation of the effects of anacetrapib through lipid-modification. https://clinicaltrials.gov/ct2/show/NCT01252953
Merki E, Graham M, Taleb A, et al. Antisense oligonucleotide lowers plasma levels of apolipoprotein (a) and lipoprotein (a) in transgenic mice. J Am Coll Cardiol. 2011;57:1611–21.
Tsimikas S, Viney NJ, Hughes SG, et al. Antisense therapy targeting apolipoprotein(a): a randomized, double-blind, placebo-controlled phase 1 study. Lancet. 2015;386:1472–83.
• Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomized, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388:2239–53. Phase II and I/IIa trials of antisense oligonucleotides targeting hepatic apolipoprotein(a) mRNA to lower Lp(a) plasma concentrations. The studies showed a 66 to 92% reduction in Lp(a) levels with the study drugs
Missala I, Kassner U, Steinhagen-Thiessen E. A systematic literature review of the association of lipoprotein(a) and autoimmune diseases and atherosclerosis. Int J Rheumatol. 2012;2012:480784.
Schultz O, Oberhauser F, Saech J, et al. Effects of inhibition of interleukin-6 signaling on insulin sensitivity and lipoprotein (a) levels in human subjects with rheumatoid diseases. PLoS One. 2010;5:e14328.
McInnes IB, Thompson L, Giles JT, et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomized, placebo-controlled study. Ann Rheum Dis. 2015;74:694–702.
Müller N, Schulte DM, Türk K, et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans. J Lipid Res. 2015;56:1034–42.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Erik Kelly declares no potential conflicts of interest.
Linda Hemphill reports personal fees from Amgen and Regeneron and grants from Regeneron/Sanofi and Ionis.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Prevention
Rights and permissions
About this article

Cite this article
Kelly, E., Hemphill, L. Lipoprotein(a): A Lipoprotein Whose Time Has Come. Curr Treat Options Cardio Med 19, 48 (2017). https://doi.org/10.1007/s11936-017-0549-z
Published:
DOI: https://doi.org/10.1007/s11936-017-0549-z