
Abstract
Recent extensive research on interleukin-2 (IL-2)/IL-2 receptor (IL-2R) biology has revealed its critical role in the regulation of immune tolerance by influencing regulatory T (Treg) cell functions and survival. Since in vivo low-dose IL-2 administration in humans has been confirmed to be safe and effective in expanding Treg, it is likely that it may be considered for the treatment of several autoimmune diseases including systemic lupus erythematousus (SLE). A recent clinical trial demonstrated the safety and efficacy of low-dose IL-2 treatment on SLE. In SLE, T cells show aberrant function such as deficient IL-2 production and abnormal signaling events. Expansion of Treg by IL-2 represents a specific strategy to control self-tolerance; however, restoration of abnormal immune function and responses should be addressed more carefully in patients with SLE considering the complexity of disease etiology and pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breakdown of immune tolerance is the critical underlying factor of autoimmunity. Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease characterized by multiple organ inflammation and immune complex formation between autoantibodies, such as anti-nuclear and anti-dsDNA, that are produced as a consequence of failure of self-tolerance and nuclear antigens [1]. The prevalence of SLE is reported to be 40–150 per 100,000 and mainly in women (9 female to 1 male). Asian, African American and Hispanic is more prevalent as compared with Caucasian [1]. The diversity of clinical manifestations has made it cumbersome to understand the pathogenesis of the disease and to develop specific drugs; a number of biological agents have failed to demonstrate clinical significance with the exception of Belimumab.
In general, control of autoreactive T cells both in the thymus and the periphery is fundamental to immune tolerance. Central tolerance is achieved in thymus, and autoreactive lymphocytes are eliminated dependent on the strength of T cell receptor (TCR) signaling. However, some T cells that can react with autoantigen escape negative selection in the thymus. Peripheral tolerance, which is responsible for keeping these self-reactive T cells inactive, is mediated by anergy or inhibition of activation. Especially, regulatory T cells (Treg) are critical for the suppression of self-reactive T cells and a hallmark of peripheral tolerance [2].
Because interleukin-2 (IL-2) is requisite for the expansion and function of Tregs, IL-2 supplementation therapy was tried first in mice with type 1 diabetes [3] and revealed that low-dose IL-2 can prevent diabetes progression with significant expansion of Tregs. Shortly thereafter, low-dose IL-2 treatment emerged for the treatment of several human autoimmune diseases [4], and demonstrated selective expansion of Treg cell populations associated with clinical response in patients with chronic graft versus host disease [5, 6], chronic hepatitis C-related vasculitis [7], type 1 diabetes (T1D) [8] and SLE [9••]. Although all types of immune cells have been reported to be involved in the development and/or progression of SLE disease manifestations, abnormalities of T lymphocyte functions including Tregs are important in the immunopathogenesis of lupus [10•, 11, 12•]. The latest clinical trial in SLE patients showed significant amelioration of disease scores with alteration of Treg/effector T cell balance. In this review, we briefly summarize the IL-2/IL-2R biology and describe the recent trials of IL-2 treatment in mice and human SLE. Lastly, we discuss the clinical significance, issues to be elucidated and therapeutic potential for future practical use.
IL-2 and IL-2 Receptor Interaction
In the decades after its identification in 1983, IL-2 has been considered an essential growth factor for lymphocytes. IL-2 is dominantly produced by activated CD4+ and CD8+ T cells and acts in an autocrine or a paracrine manner through its high-affinity receptor IL-2Rα (CD25) induced on these cells, and is required for their clonal expansion. Other IL-2Rs, IL-2Rβ (CD122), andcommon gamma chain (γc, CD132) are necessary for the transduction of IL-2 signaling [2]. In steady state, trimeric high-affinity IL-2R including CD25 (Kd = 10-11M) is mainly expressed on CD4+ Treg and dimeric low-affinity IL-2R composed of CD122 and CD132 (Kd = 10-9 M) is consistently expressed on CD8+ and NK cells and weakly on naïve CD4+ T cells. High-dose IL-2 therapy in melanoma and renal cell carcinoma has been used for more than 20 years in anticipation of the expansion of anti-cancer lymphocytes and natural killer cells that are activated and expressed CD25. It is also well established that IL-2 is essential for the maintenance and expansion of Treg cells because mice deficient in IL-2, CD25 and CD122 develop systemic auto-immunity where Treg cells are mostly absent [13]. Therefore, it is conceivable that the efficacy of IL-2 on cancers is limited because of its double-edged effect. IL-2 is also essential for driving the sensitivity of T cells to activation-induced cell death (AICD) to eliminate excessively reactive T cells, by up-regulation of Fas ligand [14]. Furthermore, IL-2 can trigger an autoregulatory feedback loop by inducing B lymphocyte-induced maturation protein (Blimp-1) expression in T cells, thereby repressing IL-2 transcription [15].
IL-2 and Responder Cells
Historically, CD8+ T cells have been thought to be the main responder cells to IL-2, and a large number of studies have argued that IL-2 controls both primary and secondary expansion of antigen-specific CD8+ T cells. IL-2 can also induce proliferation of naïve CD8+ T cells and generate effector-like T cells [2]. NK cells can proliferate and increase cytotoxic activity by IL-2, but IL-12 has a dominant effect compared with IL-2 in NK cell function. Aside from the role of IL-2 in Treg cell survival, competence and stability, IL-2 has a critical role in the development and differentiation of T cells. IL-2 promotes the development of T helper 1 (TH1), TH2 and TH9 cells [4]. By contrast, IL-2 suppresses the differentiation of CD4+ T cells into TH17 cells [16] and T follicular helper (TFH) cells [17] by a STAT5-dependent mechanism. Moreover, IL-2 can expand CD4+ PD-1+ CXCR5 + Foxp3+ follicular regulatory cells (TFR) and regulate the balance between TFH and TFR leading to the suppression of germinal center formation [18, 19]. B cells [20], group 2 innate lymphoid cells (ILC2) [21, 22], eosinophils [23], fibroblasts [24] and pulmonary endothelial cells [25] can all respond to IL-2. Although functional significance in non-lymphoid cells remains to be established, IL-2 is thought to activate and proliferate them to some degree.
IL-2-IL-2R Responsiveness
IL-2-induced oligomerization of IL-2R initiates signal transduction by recruiting janus kinase 1 (JAK1), JAK3 and activator of transcription 5 (STAT5). Responsiveness of each lymphocyte subset is well documented in cells from type 1 diabetes patients [26]. Phosphorylation of STAT5 is most sensitive in Treg cells. Only 3–7 pM (approximately 1 IU/ml) of IL-2 is sufficient for 50 % induction of pSTAT5 in Treg, whereas 100 pM is required in memory CD4+ T cells. Naïve CD4+ T cells, CD8+ T cells and NK cells require much higher concentration for substantial induction of pSTAT5. Thus, half-maximal effective concentration (EC50) of IL-2 in Treg is 10–100 times lower than that in non-Treg [27]. The higher sensitivity of Tregs to IL-2 is not solely due to their higher expression of CD25. In vitro-activated CD4+ T cells expressing high levels of CD25 are still less responsive to IL-2 than Tregs. Importantly, the Malek laboratory has found that high-protein phosphatase 2A (PP2A) activity was observed in Treg [26]. Serine and threonine residues of IL-2Rβ, JAK3 and STAT5 are de-phosphorylated by PP2A, which results in higher tyrosine phosphorylation of them upon stimulation [28]. Furthermore, Apostolidis et al. recently reported the essential role of PP2A in the function of Treg [29]. Mice that have Treg-specific ablation of PP2A develop severe autoimmune disease with an extensive repertoire of autoantibodies. The number of CD4+CD25+Foxp3+ “Treg phenotype” cells in the periphery produced IL-2 and IL- 17 but failed to display suppressive function. The study demonstrated that PP2A is requisite for Treg function because it suppresses mTORC1 activity. PP2A is also reported to be involved in the function of pulmonary endothelial cells [30, 31]. Activity of PP2A and IL-2R phosphorylation status should be determined to explain the IL-2 responsiveness of CD25+ pulmonary endothelial cells, which can cause vascular leak syndrome in high-dose IL-2 treatment but not in low-dose [25].
IL-2 Deficiency in SLE T Cells
It is well known that IL-2 production of T cells is impaired in SLE [10•]. The mechanism of IL-2 deficiency in SLE T cells has recently been extensively analyzed [11]. Lupus T cells have enhanced early CD3/TCR-mediated signaling events including heightened calcium responses leading to increased activation and enhanced translocation to the nucleus of transcription factor such as NFAT [32]. This alteration is in part due to the “rewired” TCR/CD3 complex. The CD3ζ chain is replaced by Fcε receptor 1γ (FcεR1γ, FcRγ) and FcRγ recruits tyrosine kinase Syk instead of Zap70. However, IL-2 production in SLE T cells is still impaired because transcriptional promoter cyclic AMP response element binding protein (CREB) is de-phosphoryrated through the excessive phosphatase PP2A activity [33]. Moreover, the inducible cAMP early repressor (ICER), an isoform of cyclic AMP response element modulator (CREM) and IL-2 transcriptional repressor, was recently reported to be increased in SLE T cells and to have a critical role in Th17 differentiation [34]. Enhanced activity of calcium calmodulin kinase IV (CaMKIV) [35] and decreased expression of serine-/arginine-rich splicing factor 1 (SRSF1) [36] have also been found to be involved in the transcriptional repression of IL-2.
Similarly to the T cell phenotypic change in IL-2-knockout mice, IL-2 deficiency in SLE T cells can influence T cell subset distribution. IFNγ-producing TH1 cells and IL- 17-producing TH17 cells are increased in peripheral organs such as skin and kidney [37], probably because the absence of IL-2 favors differentiation and expansion of these populations [38]. Regulatory T cell numbers are decreased in lupus-prone, and some reports in human SLE claimed decreased Treg cell numbers and function. The reduction of Treg number in patients with SLE is marred by conflicting results probably confounded by ongoing therapies and disease activation status [39]. However, a recent report indicates that functionally active CD25hi Treg cells are significantly decreased in SLE patients, which reflects an IL-2-deprived condition [40••]. CD8+ T cells and NK cells in SLE are also known to have impaired cyototoxic function that is associated with disease activity, and IL-2 restores their function [41].
IL-2 Treatment in Lupus-Prone Mice
The first report of IL-2 treatment for lupus was described in 1990, before Tregs were discovered [42]. They used IL-2-encoded vaccinia virus to introduce IL-2 in vivo and showed prolonged survival and reduced lymphadenopathy and renal damage in MRL/lpr mice. TCRαβ+CD4-CD8- (double-negative, DN) T cells, which are the most likely culprit of lymphoadenopathy in MRL/lpr mice, are consistently decreased by IL-2 treatment in all other reports (Table 1) [43–45]. Although they are also expanded in human SLE, the origin of DN T cells is still unclear. DN T cells favorably produce IL-17 in mice and humans [45, 46], indicating their involvement in the pathogenesis of SLE. By IL-2 supplmentation, Tregs were substantially increased in lymphoid organs and periphery both in NZB/NZW F1 mice and MRL/lpr mice [47]; however, we recently demonstrated that DN T cell numbers were reduced after specific expansion of cytotoxic lymphocytes using IL-2/anti-IL-2 complexes (see below) but not by IL-2/anti-IL-2 complexes directed to Tregs, suggesting the effect of IL-2 in non-Treg population for the reduction of disease progression [45].
Low-Dose IL-2 Therapy for Human Autoimmune Diseases
In high-dose IL-2 therapy in melanoma and renal cell carcinoma, 50 million IU (MIU) of IL-2 are injected intravenously every 8 hour for 15 times and the cumulative dosage was up to 3000 MIU. The response rate is less than 10 % and severe adverse effects were observed including vascular leak syndrome, kidney and liver damages [4]. Several number of preclinical studies demonstrated that low-dose IL-2 administration can selectively expand Treg cells in mice and humans. Supplementation of low-dose IL-2 ameliorates graft versus host disease and hepatitis C virus (HCV)-associated vasculitis, where the treatment augments the number of Treg cells in the periphery. In GVHD, 0.3–3 MIU of IL-2 were injected subcutaneously daily and the cumulative dosage of IL-2 was 32–320 MIU. Treg increase was 8 times higher than controls and NK cells were 2 times higher [5, 48]. For HCV-associated vasculitis, 1.5–3 MIU were injected subcutaneously daily for 5 days for 4 cycles and up to 50 MIU was used. Low-dose IL-2 clinical trials for T1D are well documented in human. Since genetic variance of IL-2, IL-2Rα and IL-2Rβ is associated with the disease susceptibility and IL-2/IL-2R impairment is observed in T1D, it is reasonable to project low-dose IL-2 as a therapeutic tool. Phase I/II trials have been conducted to optimize dosage, and 0.3–3 M IL-2 with 5 days of treatment resulted in transient upregulation of Tregs and NK with mild to moderate side effects at higher doses, without any changes with insulin C-peptide levels [8]. Although more studies are needed to define optimal treatment schemes for low-dose IL-2 it has been suggested that no more than 3 MIU per day should be administered and the cumulative doses should be less than 100 MIU [49]. Further clinical trials are now ongoing for the treatment of T1D.
Low-Dose IL-2 Clinical Trials in SLE
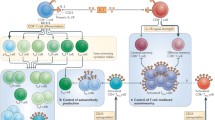
Since the decreased production of IL-2 in SLE patients likely contributes to various immune defects including reduced Treg, decreased AICD and decreased CTL responses, low-dose IL-2 treatment can restore these pathogenic mechanisms [50]. Humrich and colleagues were the first to report a case of low-dose IL-2 therapy for a patient with active SLE (Table 2) [51••]. Subcutaneous injection of 1.5–3 MIU IL-2 (aldesleukin) on 5 consecutive days resulted in the disappearance of skin eruption, myositis and serum anti-dsDNA antibodies. The SLE Disease Activity Index (SLEDAI) score decreased significantly from 14 to 4, and glucocorticoids could be reduced from 30 to 10 mg/day. The percentage of CD4+CD25+Foxp3+CD127lo Treg among CD4+ T cells was transiently upregulated at more than 40 %. Next, they started a combined phase I/IIa clinical trial addressing the safety, tolerability, efficacy and immunological responses of low-dose IL-2 therapy in patients with active and refractory SLE (PRO-IMMUN, EudraCT-number: 2013-001599-40, Germany) [40••]. In this study, Tregs from SLE patients displayed lower CD25 levels, and co-culture of CD4+ T cells with peripheral blood mononuclear cells showed deficient ex vivo IL-2 production in immune cells from SLE patients. After five patients were treated with daily subcutaneous injections of IL-2 at 1.5 MIU for 5 days, they confirmed that IL-2 therapy induced substantial increases of Tregs without major side effects. Although the study is still ongoing and not yet fully analyzed, they mentioned that the therapy showed efficacy on several organ manifestations such as skin, arthritis, myositis and alopecia. The latest clinical trial of low-dose IL-2 for 38 SLE patients was conducted in China (NCT02084238) and demonstrated that IL-2 treatment significantly decreased SLEDAI after 12 weeks [9••]. One MIU IL-2 was administered alternate days 7 times at 3 cycles. More than 80 % of patients achieved a SLE response index (SRI) with a 4-point drop in SLEDAI, with increased Tregs, and decreased TH17, TFH and DN T cells. Moreover, a clinical study of the phase II induction of Tregs by low-dose IL-2 in autoimmune and inflammatory diseases (TRANSREG) is now in progress, which will assess the safety and biological efficacy of low-dose IL-2 as a Treg inducer in 11 autoimmune and auto-inflammatory diseases including SLE (Table 2) [52]. Taken together, low-dose IL-2 treatment in SLE patients could mitigate the severity of diseases through altering the balance of T cell subsets to the decline of pathogenic immune responses (Fig. 1).

Possible mechanisms of IL-2 for the suppression of pathogenic immune responses in SLE. IL-2 administration expands Treg including TFR. IL-2, and Tregs could suppress the differentiation of TH17 and TFH cells, which induce tissue inflammation and autoantibody production, respectively. IL-2 could also influence CTL functions that could modulate DN T-cells, an aberrant population involved in tissue damage. Arrows indicate increase and/or activation of cell subset. Dotted stop lines indicate a decrease of cell subset with direct or indirect effect (undetermined at least in vivo). Treg; regulatory T-cells, T FR ; follicular regulatory T-cells, T H 17; IL-17-producing helper T-cells, T FH ; follicular helper T-cells, CTL; cytotoxic lymphocytes, DN T cells; CD3 + CD4-CD8- double-negative T-cells
Future Perspectives of IL-2 Therapy in Lupus
Even though low-dose IL-2 can substantially expand Tregs, frequent injection is required for the induction of effectiveness because IL-2 as a very short half-life in human serum (5–7 min). To overcome this disadvantage, modified IL-2 such as polyethylene glycol-modified IL-2 (PEG-IL-2), which will prolong IL-2 half-life and show effect sfor longer period, can be considered. PEG-IL-2, already constructed in the 1990s and which has undergone phase I/II clinical trials in cancer patients [53], was recently revisited and tried in mice with asthma [54]. IL-2/anti-IL-2 complexes can also prolong the half-life of IL-2. In mice, IL-2/anti-IL-2 complexes have been well analyzed: IL-2/JES6-1 specifically binds to CD25 and IL-2/S4B6 to CD122. And IL- 2/JES6-1 and IL-2/S4B6 induce specific expansion of Treg and cytotoxic lymphocytes, respectively. When IL-2/anti-IL-2 complexes for human are fully developed, they will be useful for the specific expansion of target cells with reduced frequency of injection [55]. Nanoscale liposomal polymeric gels (nanolipogels) are biologically compatible and in vivo slowly biodegradable agents. Talek and colleagues recently developed nanolipogel-encapsulated recombinant IL-2 and TGFβ inhibitor and performed intratumor injections [56]. As a result, peritumor cytotoxic lymphocytes were increased and tumor size was decreased. Furthermore, Otomo et al. showed that anti-CD4-labeled nanoparticles with CaMKIV inhibitor KN93 selectively bound to CD4+ lymphocytes regulated their functions and differentiation leading to the amelioration of disease pathology both in mice lupus and in experimental autoimmune encephalomyelitis (EAE) [57]. Usage of IL-2- nanoparticles labeled with the target cell antibody will be more specific, have longer effects and low toxicity. Furthermore, stimulation of SLAMF3, one of the susceptibility genes for SLE, has been reported to induce CD25 to facilitate the proliferation of Tregs. Therefore, the SLAMF3 agonistic agent can be a supportive drug for IL-2 treatment [58].
Conclusion
Low-dose IL-2 treatment for SLE could be a promising, selective therapeutic strategy. Since Treg is a high IL-2-sensitive lymphocyte and a critical regulator of peripheral tolerance, most of the clinical studies for autoimmune diseases are focused on the induction of Tregs. However, IL-2 could influence cells other than Tregs, such as CD8 and NK cells. The latest clinical trial showed that the DN T cell number was decreased by IL-2 treatment, indicating a distinct effect of IL-2 on the regulation of T cell populations from the expansion of Tregs. Further analysis is required to elucidate the mechanisms by which IL-2 can modulate T cell populations. Moreover, in-depth evaluation is required to determine whether up-regulation of Tregs is sufficient to deliver clinical improvement. The significance and the repercussions of low-dose IL-2 on cytotoxic lymphocytes should also be considered and addressed.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365(22):2110–21.
Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–90.
Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity. 2009;30(2):204–17.
Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol. 2015;15(5):283–94.
Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea 3rd EP, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365(22):2055–66.
Koreth J, Kim HT, Jones KT, Lange PB, Reynolds CG, Chammas MJ, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft- versus-host disease. Blood. 2016;128(1):130–7.
Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365(22):2067–77.
Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013;1(4):295–305.
•• He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4+ T cell subsets in patients with systemic lupus erythematosus. Nat Med. 2016;22(9):991–3. The newest clinical trial of low-dose IL-2 treatment for 38 SLE patients.
• Lieberman LA, Tsokos GC. The IL-2 defect in systemic lupus erythematosus disease has an expansive effect on host immunity. J Biomed Biotechnol. 2010;2010:740619. This paper summarizes mechanisms of IL-2 deficiency in SLE and its pathogenic roles for the disease progression.
Moulton VR, Tsokos GC. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest. 2015;125(6):2220–7.
• Comte D, Karampetsou MP, Tsokos GC. T cells as a therapeutic target in SLE. Lupus. 2015;24(4-5):351–63. This paper describes T cell abnormalities, cytokine aberrations in SLE patients and related theraputic options.
Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–79.
Arakaki R, Yamada A, Kudo Y, Hayashi Y, Ishimaru N. Mechanism of activation-induced cell death of T cells and regulation of FasL expression. Crit Rev Immunol. 2014;34(4):301–14.
Martins GA, Cimmino L, Liao J, Magnusdottir E, Calame K. Blimp-1 directly represses Il2 and the Il2 activator Fos, attenuating T cell proliferation and survival. J Exp Med. 2008;205(9):1959–65.
Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26(3):371–81.
Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36(5):847–56.
Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17(8):975–82.
Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. 2011;17(8):983–8.
Le Gallou S, Caron G, Delaloy C, Rossille D, Tarte K, Fest T. IL-2 requirement for human plasma cell generation: coupling differentiation and proliferation by enhancing MAPK-ERK signaling. J Immunol. 2012;189(1):161–73.
Roediger B, Kyle R, Tay SS, Mitchell AJ, Bolton HA, Guy TV, et al. IL-2 is a critical regulator of group 2 innate lymphoid cell function during pulmonary inflammation. J Allergy Clin Immunol. 2015;136(6):1653. 63 e1-7.
Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464(7293):1367–70.
Simon HU, Plotz S, Simon D, Seitzer U, Braathen LR, Menz G, et al. Interleukin-2 primes eosinophil degranulation in hypereosinophilia and Wells’ syndrome. Eur J Immunol. 2003;33(4):834–9.
Kang R, Tang D, Lotze MT, Zeh Iii HJ. Autophagy is required for IL-2- mediated fibroblast growth. Exp Cell Res. 2013;319(4):556–65.
Krieg C, Letourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci U S A. 2010;107(26):11906–11.
Yu A, Snowhite I, Vendrame F, Rosenzwajg M, Klatzmann D, Pugliese A, et al. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes. 2015;64(6):2172–83.
Tang Q. Therapeutic window of interleukin-2 for autoimmune diseases. Diabetes. 2015;64(6):1912–3.
Ross JA, Cheng H, Nagy ZS, Frost JA, Kirken RA. Protein phosphatase 2A regulates interleukin-2 receptor complex formation and JAK3/STAT5 activation. J Biol Chem. 2010;285(6):3582–91.
Apostolidis SA, Rodriguez-Rodriguez N, Suarez-Fueyo A, Dioufa N, Ozcan E, Crispin JC, et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat Immunol. 2016;17(5):556–64.
Tar K, Csortos C, Czikora I, Olah G, Ma SF, Wadgaonkar R, et al. Role of protein phosphatase 2A in the regulation of endothelial cell cytoskeleton structure. J Cell Biochem. 2006;98(4):931–53.
Kasa A, Czikora I, Verin AD, Gergely P, Csortos C. Protein phosphatase 2A activity is required for functional adherent junctions in endothelial cells. Microvasc Res. 2013;89:86–94.
Kyttaris VC, Zhang Z, Kampagianni O, Tsokos GC. Calcium signaling in systemic lupus erythematosus T cells: a treatment target. Arthritis Rheum. 2011;63(7):2058–66.
Juang YT, Wang Y, Solomou EE, Li Y, Mawrin C, Tenbrock K, et al. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J Clin Invest. 2005;115(4):996–1005.
Yoshida N, Comte D, Mizui M, Otomo K, Rosetti F, Mayadas T, et al. ICER is requisite for Th17 differentiation. Nat Commun. 2016;7:12993. doi:10.1038/ncomms12993.
Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. CaMK4-dependent activation of AKT/mTOR and CREM-alpha underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124(5):2234–45.
Moulton VR, Grammatikos AP, Fitzgerald LM, Tsokos GC. Splicing factor SF2/ASF rescues IL-2 production in T cells from systemic lupus erythematosus patients by activating IL-2 transcription. Proc Natl Acad Sci U S A. 2013;110(5):1845–50.
Crispin JC, Tsokos GC. IL-17 in systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010:943254.
Gomez-Martin D, Diaz-Zamudio M, Crispin JC, Alcocer-Varela J. Interleukin 2 and systemic lupus erythematosus: beyond the transcriptional regulatory net abnormalities. Autoimmun Rev. 2009;9(1):34–9.
Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol. 2015;45(2):344–55.
•• von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis. 2016;75(7):1407–15. The first phase I/IIa clinical trial of low-dose IL-2 for SLE patients.
Puliaeva I, Puliaev R, Via CS. Therapeutic potential of CD8+ cytotoxic T lymphocytes in SLE. Autoimmun Rev. 2009;8(3):219–23.
Gutierrez-Ramos JC, Andreu JL, Revilla Y, Vinuela E, Martinez C. Recovery from autoimmunity of MRL/lpr mice after infection with an interleukin- 2/vaccinia recombinant virus. Nature. 1990;346(6281):271–4.
Gutierrez-Ramos JC, Andreu JL, Marcos MA, Vegazo IR, Martinez C. Treatment with IL2/vaccinia recombinant virus leads to serologic, histologic and phenotypic normalization of autoimmune MRL/lpr-lpr mice. Autoimmunity. 1991;10(1):15–25.
Huggins ML, Huang FP, Xu D, Lindop G, Stott DI. Modulation of autoimmune disease in the MRL-lpr/lpr mouse by IL-2 and TGF-beta1 gene therapy using attenuated Salmonella typhimurium as gene carrier. Lupus. 1999;8(1):29–38.
Mizui M, Koga T, Lieberman LA, Beltran J, Yoshida N, Johnson MC, et al. IL-2 protects lupus-prone mice from multiple end-organ damage by limiting CD4-CD8- IL-17-producing T cells. J Immunol. 2014;193(5):2168–77.
Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181(12):8761–6.
Humrich JY, Morbach H, Undeutsch R, Enghard P, Rosenberger S, Weigert O, et al. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proc Natl Acad Sci U S A. 2010;5(179):204–9.
Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med. 2013;5(179):179ra43.
Kosmaczewska A. Low-dose interleukin-2 therapy: a driver of an imbalance between immune tolerance and autoimmunity. Int J Mol Sci. 2014;15(10):18574–92.
Humrich JY, and Riemekasten G. Restoring regulation - IL-2 therapy in systemic lupus erythematosus. Expert Rev Clin Immunol. 2016:1–8. doi:10.1080/1744666X.2016.1199957.
•• Humrich JY, von Spee-Mayer C, Siegert E, Alexander T, Hiepe F, Radbruch A, et al. Rapid induction of clinical remission by low-dose interleukin-2 in a patient with refractory SLE. Ann Rheum Dis. 2015;74(4):791–2. The first case report of low-dose IL-2 treatment for SLE.
Koreth J, Ritz J, Tsokos G, Pugliese A, Malek T, Rosenzwajg M, et al. Low-dose interleukin-2 in the treatment of autoimmune disease. Hematol Oncol Rev. 2014;10(2):157–63.
Menzel T, Schomburg A, Korfer A, Hadam M, Meffert M, Dallmann I, et al. Clinical and preclinical evaluation of recombinant PEG-IL-2 in human. Cancer Biother. 1993;8(3):199–212.
Wu K, Ma J, Bai W, Cui X, Han T, Wang S, et al. Short-term intratracheal use of PEG-modified IL-2 and glucocorticoid persistently alleviates asthma in a mouse model. Sci Rep. 2016;6:31562.
Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: biology, design and application. Trends Immunol. 2015;36(12):763–77.
Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, et al. Combination delivery of TGF- beta inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater. 2012;11(10):895–905.
Otomo K, Koga T, Mizui M, Yoshida N, Kriegel C, Bickerton S, et al. Cutting edge: nanogel-based delivery of an inhibitor of CaMK4 to CD4+ T cells suppresses experimental autoimmune encephalomyelitis and lupus-like disease in mice. J Immunol. 2015;195(12):5533–7.
Comte D, Karampetsou MP, Kis-Toth K, Yoshida N, Bradley SJ, Mizui M, et al. Engagement of SLAMF3 enhances CD4+ T-cell sensitivity to IL-2 and favors regulatory T-cell polarization in systemic lupus erythematosus. Proc Natl Acad Sci USA. 2016;113(33):9321–6.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Drs. Mizui and Tsokos declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Systemic Lupus Erythematosus
Rights and permissions
About this article

Cite this article
Mizui, M., Tsokos, G.C. Low-Dose IL-2 in the Treatment of Lupus. Curr Rheumatol Rep 18, 68 (2016). https://doi.org/10.1007/s11926-016-0617-5
Published:
DOI: https://doi.org/10.1007/s11926-016-0617-5