
Abstract
More than two decades of research has revealed a combination of proteases that determine cartilage degradation in osteoarthritis. These include metalloproteinases, which degrade the major macromolecules in cartilage, aggrecan and type II collagen, serine proteases, and cysteine proteases, for example cathepsin K. This review summarizes the function of proteases in osteoarthritis progression, as revealed by studies of genetically engineered mouse models. A brief overview of the biochemical characteristics and features of several important proteases is provided, with the objective of increasing understanding of their function. Published data reveal at least three enzymes to be major targets for osteoarthritis drug development: ADAMTS-5, MMP-13, and cathepsin K. In surgical models of osteoarthritis, mice lacking these enzymes are protected from cartilage damage and, to varying degrees, from bone changes. In-vivo studies targeting these proteases with selective small-molecule inhibitors have been performed for a variety of animal models. Mouse models will provide opportunities for future tests of the therapeutic effect of protease inhibitors, both on progression of structural damage to the joint and on associated pain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Enzymes which hydrolyze peptide bonds form 1.7 % of the human genome and are one of the largest protein families in the human body, second only to the ubiquitin ligase family [1]. There are more than 600 proteases in the human “degradome” (the complete set of proteases in an organism [1]), and they are active in almost every biological pathway of the cell cycle, wound healing, immune response, blood coagulation, and other physiological and pathological processes. Proteases often act in tightly controlled networks or cascades, and dysregulation of their activity underlies many diseases, including cancer, neurodegenerative and cardiovascular disease, and arthritis [2]. They are, therefore, possible targets for drug development. Some of the most successful drugs on the market are protease inhibitors [3]; captopril, for example, an anti-hypertensive drug targeting the metalloprotease angiotensin-converting enzyme, has been available since 1981. In 2010 it was estimated that 5–10 % of all drugs in development targeted proteases [3].
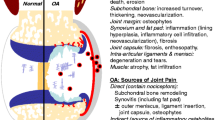
Osteoarthritis (OA), the most common form of arthritis affecting the knee, hip, and hand, is characterized by biochemical and cellular changes to all joint tissues, including cartilage, subchondral bone, synovium, meniscus, ligaments, and fat pad [4]. Characteristic of these pathological changes is altered turnover of connective tissue: subchondral bone sclerosis, bone cysts, osteophytes, synovial inflammation, fibrosis, and, most of all, loss of articular cartilage. Proteases have therefore received much attention as targets for the development of drugs to halt structural damage to the joint. Such drugs are not yet on the market.
In this review, we focus on a network of key proteases active in OA. We provide a basic overview of their biochemical characteristics and discuss insights from mouse models regarding their function in OA pathology. Studies of genetically engineered mouse models (GeMMs) reveal three proteases to be possible targets for OA therapy: ADAMTS-5, MMP-13, and cathepsin K (catK). Progress in developing selective inhibitors of these enzymes will be briefly reviewed.
The Network of Proteases Active in Osteoarthritis
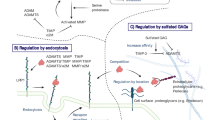
Progressive loss of articular cartilage via enzyme degradation of the extracellular matrix (ECM) is central to OA pathology (proteases in OA cartilage are reviewed in detail in Ref. [5]). Focus has long been on metalloproteinases, which are responsible for degradation of major macromolecules in cartilage, aggrecan and type II collagen (CII) (Fig. 1). Studies investigating the effect of wide-range protease inhibitors on cytokine-stimulated cartilage degradation in vitro revealed that metalloproteinases and serine and cysteine proteases are all involved in these pathways [6–8]. Figure 1 summarizes the major proteases implicated in cartilage degradation, illustrating the important function of serine, metallo, and cysteine proteases. Most protease research has focused on cartilage degradation. Cartilage loss is visible on radiology as joint-space narrowing, which is the variable by which OA progression is monitored and the primary endpoint for evaluating efficacy of disease-modifying OA drugs (DMOADs) [9]. It is not entirely understood how changes to articular cartilage are related to changes to meniscus, subchondral bone, and synovium (recently discussed in Ref. [10•]). Proteases can be produced by tissue other than cartilage, and their degradation products may affect all joint tissues. It is therefore important to study proteases in vivo, and GeMMs are a valuable tool for investigating protease function in OA pathology and progression.

Network of proteases in OA cartilage degradation. Key proteases from different classes are shown, mainly on the basis of ex-vivo human and bovine cartilage explant studies. ADAMTS-4/5 and MMP-13 are the main metalloproteases responsible for degrading aggrecan and type II collagen, respectively [79, 80]. These enzymes can also act on other substrates, degradation of which may contribute to weakening of the cartilage matrix [81]. These metalloproteases are synthesized as catalytically inactive zymogens, and it has been shown that the proforms of ADAMTS-4/5 are activated by proprotein convertases (PC) [82–84], with PACE4 identified as the major PC in human OA cartilage [78]. Tissue inhibitor of metalloprotease (TIMP)-3 is an endogenous inhibitor of MMP-13 [85] and ADAMTS-4/5 [86]. The cysteine protease, cathepsin K (catK), can also degrade collagen and aggrecan [87, 88]. Both ADAM8 [89] and HtrA1 (high temperature requirement A1) [90] can cleave fibronectin, and fibronectin fragments induce chondrocytes to release catabolic enzymes [91–93]. HtrA1 can cleave many substrates in vitro, including aggrecan, fibromodulin, and decorin [94, 95]. The function of aspartyl proteases has not yet been reported, although it was recently shown that selective inhibition of the membrane-anchored aspartyl protease, BACE-1 (beta-secretase) (primarily known for its function in Alzheimer’s disease via cleavage of amyloid precursor protein) blocked cytokine-induced aggrecan loss from bovine and human cartilage explants. The mechanism of action is not yet understood [96]
Biochemical Characteristics of OA Proteases
A brief overview of the biochemical features of the major proteases shown in Fig. 1 is provided, illustrating their function, how they cleave matrix proteins, and the rationale and methods for developing inhibitors.
Domain Structure
Domain structure analysis of ADAMTS-4, ADAMTS-5, ADAM-8, MMP-13, catK, and PACE-4 (Fig. 2) reveals that they vary greatly in length, from approximately 330 to 970 amino acids (catK and PACE4, respectively). Smaller enzymes, including catK and MMP-13, have fewer ancillary domains. CatK has a propeptide domain and a peptidase C1 domain related to papain. Its active site contains cysteine, histidine, and asparagine, which make up its catalytic triad. Unlike the other enzymes, MMP-13 has a short propeptide domain, but has a typical metallopeptidase domain (according to the Pfam database: pfam.sanger.ac.uk) and a C-terminal hemopexin domain. ADAMTS-4 and ADAMTS-5 contain a propeptide and a catalytic domain, an ADAMTS-spacer 1 domain, and at least one thrombospondin type 1 repeat. ADAM-8 has three other domains: a blood coagulation inhibitor and/or disintegrin segment, a cysteine-rich domain and an epidermal-growth-factor-like domain. PACE4 contains a galactose-binding-like domain, growth factor and/or receptor domains, a pro region, and a subtilisin domain with aspartic acid, histidine, and serine residues as catalytic triad. Catalytic domain size is 200–250 residues for all enzymes in Fig. 2 except PACE4, which has 340 residues. Larger enzymes usually have more ancillary domains, which are important for matrix location, substrate specificity, cell receptor signaling, and stability.

Domain structures of ADAMTS-4, ADAMTS-5, ADAM-8, MMP-13, cathepsin K, and PACE4. Pro, DG, Cys, EGF, and GBD indicate propeptide, blood coagulation inhibitor and/or disintegrin, ADAM and/or cysteine-rich, epidermal-growth-factor-like and galactose-binding-like domains, respectively. The histidine-sites chelating zinc ion observed for ADAMTS-4, ADAMTS-5, ADAM-8, MMP-13, and the catalytic triad residues for cathepsin K and PACE4 are marked in blue. Sequence features were retrieved from the InterPro database of EMBL-EBI, and accessions used include P29122 (PACE-4), P43235 (Cathepsin K), P78325 (ADAM-8), P45452 (MMP-13), Q9UNA0 (ADAMTS-5), and O75173 (ADAMTS-4). This graph was prepared by use of DOG [97]
Catalysis Mechanism
All serine proteinases have a similar catalytic triad of serine, histidine, and aspartic acid. Serine protease PACE4 has the catalytic triad Asp205, His246, and Ser420. Catalytic reaction by PACE4 has several steps, shown in Fig. 3a. First, the hydroxyl oxygen of Ser420 loses its hydrogen to His246. The nucleophilic oxygen targets the scissile carbonyl bond, forming the tetrahedral intermediate (Fig. 3d). Then the peptide bond is cleaved (as indicated by the arrows), and the N-terminal portion of the substrate diffuses away. The remaining substrate has a temporary covalent bond with Ser420, forming an acyl-enzyme intermediate. A water molecule targets the ester bond of this acyl-enzyme intermediate, forming a second tetrahedral intermediate, and finally this tetrahedral intermediate disassembles, releasing the C-terminal portion of the substrate.

Schematic diagrams of the catalytic mechanism for serine, cysteine, and metallo-proteinases. The numbering of the residues involved in a catalytic reaction is in accord with PACE4 (serine proteinase, a and d), cathepsin K (cysteine proteinase, b and e) and ADAMTS-5 (metalloproteinase, c and f), respectively
The catalytic mechanism of cysteine proteinase is very similar to that of serine proteinases, but with a mercapto sulfur of cysteine as nucleophilic group (Fig. 3b, e). The catalytic triad of catK is Cys139, His276, and Asn296.
The catalytic mechanism of metalloproteinases differs from that of serine and cysteine proteinases. Two mechanisms have been proposed, the promoted-water pathway and nucleophile pathway [11]. Figures 3c, f show the promoted-water pathway for ADAMTS-5. First, zinc-coordinated water loses a hydrogen atom to Glu411 and the nucleophilic oxygen of water targets the scissile carbonyl bond, forming a tetrahedral intermediate (as indicated by arrows in Figs. 3c and f). In this state, zinc links to the oxygen in the carbonyl substrate instead of the oxygen in water. Next, as the arrows indicate, the peptide bond is cleaved and the N-terminal portion of the substrate lost. The remaining C-terminal portion of the substrate forms an enzyme–product complex. Finally, the product is released from the enzyme, enabling the next catalytic cycle to start.
Active Site Structures
With the exception of PACE4, many catalytic domain structures of the above proteases have been determined (Fig. 4). The catalytic domains of ADAM-8, ADAMTS-4, ADAMTS-5, and catK have similar sequence lengths and are therefore of similar size. MMP-13 has a smaller catalytic domain than the other proteases. A homology model reveals that, because it contains the greatest number of amino acids, PACE4 has the largest catalytic domain. Secondary structures and patterns of ADAM-8, ADAMTS-4, and ADAMTS-5 are similar (Figs. 4a–c). MMP-13 (Fig. 4d) has a different structural pattern; however, the active sites of all four metalloproteinases are similar and use the same mechanism of peptide hydrolysis (Fig. 3). ADAM-8, ADAMTS-4, and ADAMTS-5 all have an α-helix, which forms the base of the active site. There is also an α-helix and several loops located at the right-hand side of the ligand. MMP-13 lacks an α-helix. CatK and PACE4 are different classes of proteinase, and their active sites are very different. Although ADAM-8, ADAMTS-4, ADAMTS-5, and MMP-13 have similar tertiary structures, their active site pockets are of different size, shape, hydrophobicity, and electrostatic potential (data not shown). Because these properties determine the proteases’ binding specificity for different compounds (ligands) [12, 13•, 14], it is possible to design potent and, more importantly, selective inhibitors for each protease, even if they are closely related. It must be taken into account that proteins are not totally rigid, adopting different conformations to accommodate different ligands [13•]. This is why one compound can bind to several target proteins, and why developing selective inhibitors targeting only one member of a family is challenging.

3D crystal structures of the catalytic domains of ADAMTS-4, ADAMTS-5, ADAM-8, MMP-13, cathepsin K, and PACE-4. Proteins are represented as ribbons, ligands as sticks, and zinc ions as gray spheres. The model of PACE4 was created by use of SWISS-MODEL [98]. PACE4A-I was chosen as the target sequence, and the crystal structure of furin (PDB code: 1P8J) [99] was used to model PACE4. a–f show the structures of ADAM-8, ADAMTS-5, ADAMTS-4, MMP-13, cathepsin K, and PACE4A-I, respectively. The catalytic triad residues of PACE4A-I are represented as sticks. The PDB codes used were 4DD8 (ADAM-8) [100], 3C9E (Cathepsin K) [101], 3ELM (MMP-13) [12], 2RJP (ADAMTS-4) [102], and 3B8Z (ADAMTS-5) [103]. The graphs were prepared by use of PyMOL [104]
Mouse Models Confirm the Importance of Proteases in OA
The mouse degradome is larger than the human degradome, particularly for proteases with immunological and reproductive functions, and lacks some important human enzymes, including MMP-1 [15]. Despite this, mouse models of OA, in particular models using knockout (KO) mice, are invaluable for target validation [16]. In this review, we focus on GeMMs and expression studies that use the most common surgical and spontaneous models of murine OA (the models are reviewed in Ref. [17]).
Validation by Use of Genetically Engineered Mouse Models (GeMMs)
Surgical Models
Most protease KO mice have been studied by use of surgical models (Table 1). OA induced by destabilization of the medial meniscus (DMM) is often chosen as a model because the slow progression of structural changes to the joint enables long-term follow-up [18]. Inhibition of cartilage degradation remains the main criterion for deciding whether KO mice are protected against OA, but an increasing number of analyses also include subchondral bone changes. The role of synovial change in these models is under active investigation. Of the eight proteases and endogenous inhibitors tested, Timp2 [19] and Mmp9 [16, 20] deficient mice are more susceptible to OA. Loss of Timp2 promotes vascular invasion of the periarticular region before OA changes develop, suggesting increased angiogenesis may drive OA progression [19]. Because MMP-9 has anti-inflammatory functions, including degradation of IL-1β, it is postulated that it has a protective function in OA [16]. Lack of Adamts4 [21], Mmp3 [16], Mmp12 [16], or Mmp17 [22] does not affect OA progression. MMP-17 (also known as MT4-MMP) was investigated regarding its possible involvement in activating ADAMTS-4 [23]. Mmp17 null mice had no protection in the DMM model but were protected from loss of glycosaminoglycan (GAG) into synovial fluid after intra-articular injection of IL-1β, suggesting that MMP-17 may be involved in inflammation-associated joint destruction [22]. Only three KO strains, Adamts5, Mmp13, and catK (Ctsk) null mice, were protected from OA development, and 100 % protection against cartilage degeneration was never reported.
Adamts5 null mice are protected from aggrecan loss and cartilage degradation after instability has been induced by DMM surgery [24]. Protection from cartilage degradation and subchondral bone remodeling up to six months after DMM has been reported, but no protection from osteophyte development was observed [24, 25]. Adamts4/5 null mice have similar protection against cartilage degeneration [26]. Two transgenic mouse strains were developed to compare the effects of MMP and ADAMTS cleavage in the interglobular domain (IGD) of aggrecan. Chloe mice are resistant to MMPs cleavage at the Asn341–Phe342 site [27], whereas Jaffa mice are resistant to ADAMTS cleavage at the Glu373–Ala374 site [28]. Chloe mice studied by use of the DMM model developed OA similar to that of WT mice, whereas Jaffa mice had cartilage degradation protection similar to that of Adamts5 null mice [24, 28]. This suggests that blocking ADAMTS-mediated cleavage in aggrecan IGD is sufficient to protect against cartilage erosion.
Mmp13 null mice were not protected from aggrecan loss after DMM, but had significantly less cartilage damage up to 8 weeks after surgery [29]. The mice developed localized proteoglycan depletion on the femoral side, but this did not result in increased structural damage: cartilage structure was preserved, revealing the importance of collagen cleavage to loss of cartilage tissue in OA [29]. In a different joint instability model, cartilage-specific Mmp13-deficient mice were protected against cartilage degeneration up to 16 weeks post surgery [30]. The mice studied were Mmp13 Col2ER mice (Mmp13 null crossed with Col2CreER transgenic); the Col2a1-Cre mouse has altered gene expression in both synovial fibroblasts and chondrocytes [31]. DMM in Mmp13-overexpressing mice resulted in accelerated cartilage degradation [16], further confirming the function of this protease in OA. Transcription factors NF-κB, HIF2α, Ihh, Runx2, ELF3, C/EBPβ, and AP-1 have been implicated in regulation of MMP-13, but it is not yet understood how each of these affects OA pathogenesis [5, 32, 33]. HIF2α-deficient mice (Epas1 +/−) and Runx2 +/− mice are protected against cartilage degradation [34] and osteophyte formation in joint instability models [35, 36]. These studies reinforce the importance of MMP-13 to OA pathogenesis; however, the factors regulating its transcription have many biological functions and are therefore unlikely to be viable drug targets.
Ctsk null mice develop osteopetrosis, with abnormally high bone mineral content and volume by two months of age [37]. At 25 weeks of age these increases were still apparent in the tibial metaphysis, but subchondral bone seemed unaffected [38•]. In two models, Ctsk null mice were protected against cartilage degradation and subchondral bone changes up to 16 weeks after surgery, although osteophytosis was not significantly reduced [38•, 39••]. Eight weeks after partial medial menisectomy, wild-type mice expressed catK in chondrocytes and synovium [38•]. Ctsk null mice showed possible protection against osteophyte formation, but this was not analyzed for statistical significance [38•].
Spontaneous OA
Mice are naturally susceptible to OA of the knee joints; individual susceptibility varies, depending on background strain [40]. Slowly-progressive natural OA in C57Bl/6 mice has been studied in detail [41, 42]. Analysis of articular cartilage integrity, chondrocyte cell death, and subchondral bone up to 23 months of age revealed joint changes comparable with those seen 5.5 months after DMM surgery, but with reduced osteophyte incidence and development [41].
Of transgenic mice studied for progression of spontaneous OA, mice deficient in Adam15 [43], Mmp2 [44], or Mmp14 [45] develop osteoarthritis more rapidly than WT mice (Table 2). Mmp3 null mice have OA similar to that of WT (B10.RIII) mice by 1 year of age, but by 2 years have significantly less cartilage degeneration. There was no significant difference in fibrosis or synovial infiltration [46]. There is as yet no report on susceptibility of Adamts4, Adamts5, or Mmp13 null mice to spontaneous OA, so it is not possible to compare their protection with that of Mmp3 null mice. Ctsk null mice have only been evaluated up to 25 weeks of age, which is not long enough for spontaneous OA to develop in matched WT mice [38•]. There are, however, data indirectly confirming the function of these proteases: mice lacking TIMP-3, an endogenous inhibitor of ADAMTS-4/5 and MMP-13, develop proteoglycan loss earlier in the aging process [47]. OA has been reported to develop more rapidly in mice over-expressing hMmp13 [48], although another group could not repeat these results [16]. Ctsk over-expressing mice develop progressive synovitis with age, resulting in synovial hyperplasia, fibrosis, and joint destruction [49].
Other Models
The function of some of these enzymes has been studied by use of acute inflammation models. Adamts5 null mice are protected from cartilage proteoglycan loss, but not from synovitis, in the acute phase (day three) of the inflammatory antigen-induced arthritis (AIA) model [50]. In the AIA model, Jaffa mice have protection from cartilage proteoglycan loss during days 1–5, but by day seven develop cartilage erosion similar to that of Chloe and WT mice, with extensive proteoglycan loss in all genotypes. Between days seven and 28, however, Jaffa mice can initiate proteoglycan restoration, resulting in significantly lower cartilage erosion scores [28]. With the results from the DMM model, these findings strongly suggest that blocking aggrecanolysis in the IGD domain may be beneficial throughout OA pathogenesis, slowing down progressive cartilage erosion and perhaps also offering the possibility of repair.
In a model of joint deterioration induced by intra-articular injection of TGF-β1 and treadmill running, Adamts5 null mice were protected from proteoglycan loss and development of fibrosis on the surface of knee joint tissues [51], suggesting that ADAMTS-5 may be involved in fibrosis.
It would seem worthwhile to use GeMM to investigate possible OA pathogenesis functions for PACE4, ADAM8, and HtrA1, the remaining proteases in the proposed scheme. Such experiments, however, are not yet in the public domain. In the collagen-induced arthritis model, transgenic mice over-expressing a catalytically inactive mutant of Adam8 had significantly less bone resorption, inflammation, and cartilage degradation than WT mice [52]. It has been postulated that HtrA1 participates in degradation of the pericellular matrix in response to biomechanical stress, exposing the receptor tyrosine kinase DDR2 to its ligand, native CII, and inducing release of MMP-13 [53]. This mechanism may be responsible for the attenuated OA progression of Ddr2 +/− mice after DMM surgery [54•]. Data on Htra1-deficient mice are not available.
Murine Expression Data
Several microarray expression studies of murine OA models were published in 2012 [55•, 56, 57]. One study of expression patterns in articular cartilage of aging STR/Ort mice, which are spontaneously susceptible to OA-like changes [57], revealed that many proteases believed to affect human OA were up-regulated in STR/Ort articular cartilage, including many of those listed in Fig. 1. The authors combined data from OA mice at 18 and 40 weeks of age and found that the protease-related genes Adam8, Adamts1, Adamts4, Ctsk, Htra1, Mmp2, Mmp3, Mmp13, Mmp14, Timp1, Timp2, and Timp3 were significantly up-regulated compared with non-OA samples, thus largely confirming the networks identified in years of research on isolated cartilage from several species, including human OA samples. Other up-regulated protease genes included Capn6 (Calpain-6), Dpp7 (dipeptidylpeptidase 7), Mmp19, and Prss23 (protease serine 23). A few genes, including Dpep1 (dipeptidase 1), and Gzma (granzyme A), were down-regulated.
Another microarray study [56] analyzed gene expression in whole-knee-joint samples taken 8 weeks post-DMM. Surgery was performed on young mice (12 weeks of age) and on 12-month old mice, which develop more severe disease, and samples from the two age groups were compared. Compared with the previously mentioned study, fewer significant changes to protease-related genes were reported. Htra1, Mmp2, Mmp3, and Timp1 were up-regulated in both young and old mice. Pcsk5, which encodes the proprotein convertase PC5/6A, was up-regulated in old mice, but not young mice.
A third study analyzed samples taken from whole-knee-joint extracts 6 h and 4 weeks post-DMM [55•]. Six hours after surgery, Timp1, Mmp3, Adamts1, Adamts4, Mmp19, and Adamts5 protease-related genes were up-regulated compared with sham mice. Timp1 and Mmp3 were still up-regulated 4 weeks post-DMM. Follow-up qRT–PCR studies found that, six hours after surgery, Adamts5 and Mmp3 were increased in cartilage, but not in meniscus or in bone. Joint immobilization by means of sciatic neurectomy, which inhibited movement in the operated limb and protected against development of OA, negated the response of selected genes—including Adamts5, Adamts1, and Adamts4—in whole-knee extracts taken 6 h post-surgery.
These expression studies of diverse murine models confirm that proteases are tightly regulated during all stages of OA (as early as six h after surgery, and up to 40 weeks of age for STR/ort mice). Such studies will enable researchers to determine the effect of proteases over time and in different tissue, and to evaluate age-related differences. It is possible that protease expression in other tissue contributes to OA pathogenesis. A study investigating the effect of a high-fat diet (HFD) on genetically identical eight-week-old male B6 mice reported highly variable obesity [58]. Micro-array and RT–PCR analysis of adipose tissue revealed that Adamts5 was over-expressed in mice which gained more weight. Independent studies revealed that “high-gainer” mice fed an HFD developed more safranin O-staining loss than “low-gainer” mice [59]. This indicates that induction of ADAMTS-5 in other tissue, possibly including tissue outside the joint, may contribute to OA development. As far as we are aware, obesity-induced OA has not been studied in Adamts5 null mice.
Proof of Concept Studies with Selective Inhibitors
Studies of GeMMs have reduced potential target proteases down to three enzymes critical for development of OA joint damage: ADAMTS-5, MMP-13, and catK.
In-vivo studies targeting these proteases with selective small-molecule inhibitors have been performed on a variety of animal models, but published information is nonetheless limited, especially for ADAMTS-5. Wyeth’s ADAMTS-4/5 inhibitor was shown to protect against cartilage degeneration and weight-bearing changes for a rat medial meniscal tear model [60], but no mouse data are available for this compound. Phase 1 clinical trials have been completed and revealed no safety concerns (NCT00427687, NCT00454298; www.clinicaltrials.gov). As a promising alternative to small molecule inhibition, GlaxoSmithKline recently reported a neutralizing anti-ADAMTS-5 antibody [61]. When administered prophylactically to mice over the course of 8 weeks post-DMM, the antibody was shown to protect against joint damage [61, 62]. The antibody has also been shown to fully penetrate mouse cartilage when administered intraperitoneally to mice 6 weeks post-DMM surgery [62]. Therapeutic efficacy of the antibody for established OA was not reported.
MMP inhibitor development has been hindered by non-specificity resulting in musculoskeletal syndrome (MSS) [63]. Studies of more selective inhibitors targeting MMP-13 have not reported these problems, and seem to offer protection from cartilage loss for a variety of species [64–66, 67••] and to prevent pain behavior in rats [64, 68]. In a recent study using a murine joint-instability model, an MMP-13 inhibitor administered at the time of surgery reduced cartilage loss up to 12 weeks follow-up [30]. Again, therapeutic procedures have not been reported, and no MMP-13 inhibitor has yet reached clinical trial.
Studies of catK inhibitors using OA models of species other than mice have reported beneficial effects, both on structural progression of disease [39••, 69] and on joint pain, on long-term spontaneous OA in Dunkin–Hartley guinea pigs [70]. Novartis recently completed three phase 2 clinical trials testing balicatib (AAE581), a catK inhibitor, for safety and efficacy for osteoporosis [71] and knee OA (NCT00170911, NCT00100607, and NCT00371670). Patients with painful knee OA (KL Grade 3) were studied by a 12-month placebo-controlled trial evaluating safety, tolerability, and disease-modifying efficacy of daily oral treatment. Results from this study have not yet been published.
Conclusions and Future Challenges
GeMMs have been used for protease gain-of-function and loss-of-function studies, but most inhibitor studies have been performed on other species. Murine models, particularly the DMM model, have the advantage of being slowly progressive and therefore enabling investigation of therapeutic efficacy at different stages of OA progression. Using these models to test compounds targeting ADAMTS-5, MMP-13, and catK will provide insight into their therapeutic efficacy for established OA. Time-course experiments may determine whether there is an optimum time to initiate pharmacological intervention, and if and when it becomes too late for treatment.
What matters to the OA patient are the symptoms, particularly pain, that accompany the joint failure characteristic of OA. It is not yet known whether halting structural progression will be accompanied by pain alleviation. Techniques to model OA pain in mice are being developed [72], and may address this gap in our understanding of OA. It was recently shown that Adamts-5-deficient mice are protected from mechanical allodynia after DMM surgery [73]. This may simply be because they are protected from developing OA in this model, but could indicate a direct effect of ADAMTS-5 on the nervous system. Adamts5 is expressed in dorsal root ganglia in adult mice [74]. Over the last decade proteases, particularly MMPs, have been increasingly implicated in mediating inflammation because of their ability to cleave cytokines, chemokines, and receptors, and to degrade ECM [75]. Pain generation requires interaction between the immune and nervous systems (reviewed in Ref. [76]). It therefore seems probable that MMPs and other proteases participate in development and maintenance of pain. A multi-cohort study strongly associated a polymorphism in the gene encoding PACE4 (PCSK6) with protection against pain from knee OA [77], and many studies report Pcsk6 null mice to be protected against pain [77]. Because PACE4 is the main activator of pro-aggrecanases in human OA cartilage [78], this could suggest common pathways for structural progression and pain mechanisms in OA.
A possible function for proteases in mouse models of OA pain has not yet been studied, but proteases may be a promising subject for research addressing the relationship between structural damage and pain.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Lopez-Otin C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. 2002;3(7):509–19.
Lopez-Otin C, Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. 2008;283(45):30433–7.
Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat Rev Drug Discov. 2010;9(9):690–701.
Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–707.
Troeberg L, Nagase H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim Biophys Acta. 2012;1824(1):133–45.
Milner JM, Rowan AD, Elliott SF, Cawston TE. Inhibition of furin-like enzymes blocks interleukin-1alpha/oncostatin M-stimulated cartilage degradation. Arthritis Rheum. 2003;48(4):1057–66.
Sondergaard BC, Henriksen K, Wulf H, Oestergaard S, Schurigt U, Brauer R, et al. Relative contribution of matrix metalloprotease and cysteine protease activities to cytokine-stimulated articular cartilage degradation. Osteoarthritis Cartilage. 2006;14(8):738–48.
Milner JM, Patel A, Rowan AD. Emerging roles of serine proteinases in tissue turnover in arthritis. Arthritis Rheum. 2008;58(12):3644–56.
Le Graverand-Gastineau MP. Disease modifying osteoarthritis drugs: facing development challenges and choosing molecular targets. Curr Drug Targets. 2010;11(5):528–35.
• Little CB, Fosang AJ. Is cartilage matrix breakdown an appropriate therapeutic target in osteoarthritis—insights from studies of aggrecan and collagen proteolysis? Curr Drug Targets. 2010;11(5):561–75. Provides a good discussion of the relationship between cartilage structural integrity and pathology in other tissues in the OA joint.
Wu S, Zhang C, Xu D, Guo H. Catalysis of carboxypeptidase A: promoted-water versus nucleophilic pathways. J Phys Chem B. 2010;114(28):9259–67.
Monovich LG, Tommasi RA, Fujimoto RA, Blancuzzi V, Clark K, Cornell WD, et al. Discovery of potent, selective, and orally active carboxylic acid based inhibitors of matrix metalloproteinase-13. J Med Chem. 2009;52(11):3523–38.
• Shieh HS, Tomasselli AG, Mathis KJ, Schnute ME, Woodard SS, Caspers N, et al. Structure analysis reveals the flexibility of the ADAMTS-5 active site. Protein Sci. 2011;20(4):735–44. First demonstration that the active site of ADAMTS-5 can undergo a conformational change induced by a small molecule; may enable the design of inhibitors with improved potency and selectivity profiles.
Tortorella MD, Tomasselli AG, Mathis KJ, Schnute ME, Woodard SS, Munie G, et al. Structural and inhibition analysis reveals the mechanism of selectivity of a series of aggrecanase inhibitors. J Biol Chem. 2009;284(36):24185–91.
Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet. 2003;4(7):544–58.
Glasson SS. In vivo osteoarthritis target validation utilizing genetically-modified mice. Curr Drug Targets. 2007;8(2):367–76.
Little CB, Smith MM. Animal Models of Osteoarthritis. Curr Rheumatol Rev. 2008;4(3):175–82.
Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15(9):1061–9.
Mi M, Shi S, Li T, Holz J, Lee YJ, Sheu TJ, et al. TIMP2 deficient mice develop accelerated osteoarthritis via promotion of angiogenesis upon destabilization of the medial meniscus. Biochem Biophys Res Commun. 2012;423(2):366–72.
Glasson S, Blanchet T, Morris EA. Less Severe OA is Observed in IL-1b KO Mice and More Severe OA is Observed in MMP-9 and MK2 KO Mice in a Surgical Model of OA. ORS 51st Annual Meeting; Feb 20–23; Washington, DC 2005. p. 0251.
Glasson SS, Askew R, Sheppard B, Carito BA, Blanchet T, Ma HL, et al. Characterization of and osteoarthritis susceptibility in ADAMTS-4-knockout mice. Arthritis Rheum. 2004;50(8):2547–58.
Clements KM, Flannelly JK, Tart J, Brockbank SM, Wardale J, Freeth J, et al. Matrix metalloproteinase 17 is necessary for cartilage aggrecan degradation in an inflammatory environment. Ann Rheum Dis. 2011;70(4):683–9.
Gao G, Plaas A, Thompson VP, Jin S, Zuo F, Sandy JD. ADAMTS4 (aggrecanase-1) activation on the cell surface involves C-terminal cleavage by glycosylphosphatidyl inositol-anchored membrane type 4-matrix metalloproteinase and binding of the activated proteinase to chondroitin sulfate and heparan sulfate on syndecan-1. J Biol Chem. 2004;279(11):10042–51.
Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434(7033):644–8.
Botter SM, Glasson SS, Hopkins B, Clockaerts S, Weinans H, van Leeuwen JP, et al. ADAMTS5−/− mice have less subchondral bone changes after induction of osteoarthritis through surgical instability: implications for a link between cartilage and subchondral bone changes. Osteoarthritis Cartilage. 2009;17(5):636–45.
Majumdar MK, Askew R, Schelling S, Stedman N, Blanchet T, Hopkins B, et al. Double-knockout of ADAMTS-4 and ADAMTS-5 in mice results in physiologically normal animals and prevents the progression of osteoarthritis. Arthritis Rheum. 2007;56(11):3670–4.
Little CB, Meeker CT, Hembry RM, Sims NA, Lawlor KE, Golub SB, et al. Matrix metalloproteinases are not essential for aggrecan turnover during normal skeletal growth and development. Mol Cell Biol. 2005;25(8):3388–99.
Little CB, Meeker CT, Golub SB, Lawlor KE, Farmer PJ, Smith SM, et al. Blocking aggrecanase cleavage in the aggrecan interglobular domain abrogates cartilage erosion and promotes cartilage repair. J Clin Invest. 2007;117(6):1627–36.
Little CB, Barai A, Burkhardt D, Smith SM, Fosang AJ, Werb Z, et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60(12):3723–33.
Wang M, Sampson ER, Jin H, Li J, Ke QH, Im HJ et al. MMP13 is a Critical Target Gene During the Progression of Osteoarthritis. Arthritis Res Ther. 2013;In Press.
Fosang AJ, Golub SB, East CJ, Rogerson FM. Abundant LacZ activity in the absence of Cre expression in the normal and inflamed synovium of adult Col2a1-Cre; ROSA26RLacZ reporter mice. Osteoarthritis Cartilage. 2013;(21):401–4.
Goldring MB, Otero M, Plumb DA, Dragomir C, Favero M, El Hachem K, et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur Cell Mater. 2011;21:202–20.
Husa M, Liu-Bryan R, Terkeltaub R. Shifting HIFs in osteoarthritis. Nat Med. 2010;16(6):641–4.
Yang S, Kim J, Ryu JH, Oh H, Chun CH, Kim BJ, et al. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;16(6):687–93.
Saito T, Fukai A, Mabuchi A, Ikeda T, Yano F, Ohba S, et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med. 2010;16(6):678–86.
Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006;54(8):2462–70.
Gowen M, Lazner F, Dodds R, Kapadia R, Feild J, Tavaria M, et al. Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res. 1999;14(10):1654–63.
• Kozawa E, Nishida Y, Cheng XW, Urakawa H, Arai E, Futamura N, et al. Osteoarthritic change is delayed in a Ctsk-knockout mouse model of osteoarthritis. Arthritis Rheum. 2012;64(2):454–64. First report on protection against osteoarthritis-like changes after destabilization of the knee in Ctsk-null mice (with 39).
•• Hayami T, Zhuo Y, Wesolowski GA, Pickarski M, le Duong T. Inhibition of cathepsin K reduces cartilage degeneration in the anterior cruciate ligament transection rabbit and murine models of osteoarthritis. Bone. 2012;50(6):1250–9. First report on reduced osteoarthritis after ACL transection for mice that lack cathepsin K; also describes an eight-week experiment with a CatK inhibitor in a rabbit ACL transection model.
Bendele AM. Animal models of osteoarthritis. J Musculoskelet Neuronal Interact. 2001;1(4):363–76.
McNulty MA, Loeser RF, Davey C, Callahan MF, Ferguson CM, Carlson CS. Histopathology of naturally occurring and surgically induced osteoarthritis in mice. Osteoarthritis Cartilage. 2012;20(8):949–56.
van der Kraan PM, Stoop R, Meijers TH, Poole AR, van den Berg WB. Expression of type X collagen in young and old C57Bl/6 and Balb/c mice. Relation with articular cartilage degeneration. Osteoarthritis Cartilage. 2001;9(2):92–100.
Bohm BB, Aigner T, Roy B, Brodie TA, Blobel CP, Burkhardt H. Homeostatic effects of the metalloproteinase disintegrin ADAM15 in degenerative cartilage remodeling. Arthritis Rheum. 2005;52(4):1100–9.
Mosig RA, Dowling O, DiFeo A, Ramirez MC, Parker IC, Abe E, et al. Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. 2007;16(9):1113–23.
Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99(1):81–92.
Blom AB, van Lent PL, Libregts S, Holthuysen AE, van der Kraan PM, van Rooijen N, et al. Crucial role of macrophages in matrix metalloproteinase-mediated cartilage destruction during experimental osteoarthritis: involvement of matrix metalloproteinase 3. Arthritis Rheum. 2007;56(1):147–57.
Sahebjam S, Khokha R, Mort JS. Increased collagen and aggrecan degradation with age in the joints of Timp3(−/−) mice. Arthritis Rheum. 2007;56(3):905–9.
Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. 2001;107(1):35–44.
Morko J, Kiviranta R, Joronen K, Saamanen AM, Vuorio E, Salminen-Mankonen H. Spontaneous development of synovitis and cartilage degeneration in transgenic mice overexpressing cathepsin K. Arthritis Rheum. 2005;52(12):3713–7.
Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434(7033):648–52.
Li J, Anemaet W, Diaz MA, Buchanan S, Tortorella M, Malfait AM, et al. Knockout of ADAMTS5 does not eliminate cartilage aggrecanase activity but abrogates joint fibrosis and promotes cartilage aggrecan deposition in murine osteoarthritis models. J Orthop Res. 2011;29(4):516–22.
Zack MD, Melton MA, Stock JL, Storer CE, Barve RA, Minnerly JC, et al. Reduced incidence and severity of experimental autoimmune arthritis in mice expressing catalytically inactive A disintegrin and metalloproteinase 8 (ADAM8). Clin Exp Immunol. 2009;158(2):246–56.
Xu L, Polur I, Servais JM, Hsieh S, Lee PL, Goldring MB, et al. Intact pericellular matrix of articular cartilage is required for unactivated discoidin domain receptor 2 in the mouse model. Am J Pathol. 2011;179(3):1338–46.
• Xu L, Servais J, Polur I, Kim D, Lee PL, Chung K, et al. Attenuation of osteoarthritis progression by reduction of discoidin domain receptor 2 in mice. Arthritis Rheum. 2010;62(9):2736–44. On the basis of studies in Ddr2−/− mice, the authors propose a chain of molecular events that underlies the process of articular cartilage degeneration, eventually leading to the development of OA.
• Burleigh A, Chanalaris A, Gardiner MD, Driscoll C, Boruc O, Saklatvala J, et al. Joint immobilization prevents murine osteoarthritis and reveals the highly mechanosensitive nature of protease expression in vivo. Arthritis Rheum. 2012;64(7):2278–88. Expression studies of the DMM model reveal rapid induction of genes encoding proteases, including aggrecanases. It is also shown that some genes, including Adamts5, are mechanosensitive.
Loeser RF, Olex AL, McNulty MA, Carlson CS, Callahan MF, Ferguson CM, et al. Microarray analysis reveals age-related differences in gene expression during the development of osteoarthritis in mice. Arthritis Rheum. 2012;64(3):705–17.
Poulet B, Ulici V, Stone TC, Pead M, Gburcik V, Constantinou E, et al. Time-series transcriptional profiling yields new perspectives on susceptibility to murine osteoarthritis. Arthritis Rheum. 2012;64(10):3256–66.
Koza RA, Nikonova L, Hogan J, Rim JS, Mendoza T, Faulk C, et al. Changes in gene expression foreshadow diet-induced obesity in genetically identical mice. PLoS Genet. 2006;2(5):e81.
Griffin TM, Fermor B, Huebner JL, Kraus VB, Rodriguiz RM, Wetsel WC, et al. Diet-induced obesity differentially regulates behavioral, biomechanical, and molecular risk factors for osteoarthritis in mice. Arthritis Res Ther. 2010;12(4):R130.
Glasson S, Bendele A, Sum PE, Tam S, Tejada J, Rivera-Bermudez M et al. Selective Aggrecanase Inhibition is Disease Modifying and Pain Alleviating in a Rat Meniscal Tear Model of Osteoarthritis. OARSI Annual Meeting; Montreal, Canada: Osteoarthritis Cartilage; 2009. p. S56.
Larkin J. Linking an ADAMTS5-specific Therapeutic Monoclonal Antibody to a Sensitive Biochemical Marker of Target Engagement and Activity for Potential Application as a Companion Diagnostic. OARSI Annual Meeting; April 26–29; Barcelona, Spain: Osteoarthritis Cartilage; 2012. p. S290.
Burden M, Hamblin P, Larkin J, White J, inventors; Glaxo Group Limited, Burden, M., Hamblin, P., Larkin, J., White, J., assignee. Polypeptides and Method of Treatment patent WO 2011/002968 A2. 2010.
Krzeski P, Buckland-Wright C, Balint G, Cline GA, Stoner K, Lyon R, et al. Development of musculoskeletal toxicity without clear benefit after administration of PG-116800, a matrix metalloproteinase inhibitor, to patients with knee osteoarthritis: a randomized, 12-month, double-blind, placebo-controlled study. Arthritis Res Ther. 2007;9(5):R109.
Baragi VM, Becher G, Bendele AM, Biesinger R, Bluhm H, Boer J, et al. A new class of potent matrix metalloproteinase 13 inhibitors for potential treatment of osteoarthritis: evidence of histologic and clinical efficacy without musculoskeletal toxicity in rat models. Arthritis Rheum. 2009;60(7):2008–18.
Johnson AR, Pavlovsky AG, Ortwine DF, Prior F, Man CF, Bornemeier DA, et al. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J Biol Chem. 2007;282(38):27781–91.
Jungel A, Ospelt C, Lesch M, Thiel M, Sunyer T, Schorr O, et al. Effect of the oral application of a highly selective MMP-13 inhibitor in three different animal models of rheumatoid arthritis. Ann Rheum Dis. 2010;69(5):898–902.
•• Settle S, Vickery L, Nemirovskiy O, Vidmar T, Bendele A, Messing D, et al. Cartilage degradation biomarkers predict efficacy of a novel, highly selective matrix metalloproteinase 13 inhibitor in a dog model of osteoarthritis: confirmation by multivariate analysis that modulation of type II collagen and aggrecan degradation peptides parallels pathologic changes. Arthritis Rheum. 2010;62(10):3006–15. Demonstrates chondroprotective effects of a potent and highly selective MMP-13 inhibitor for a canine OA model. Biomarkers of cartilage degradation are also evaluated.
Black RA, Gabel C, Lively S, Toteva M, Fan P, Tocker J et al. MMP-13 Inhibitors Reduce Nociception in a Rat Model of Osteoarthritis. OARSI Annual Meeting; Brussels, Belgium: Osteoarthritis Cartilage; 2010. p. S25.
Connor JR, LePage C, Swift BA, Yamashita D, Bendele AM, Maul D, et al. Protective effects of a cathepsin K inhibitor, SB-553484, in the canine partial medial meniscectomy model of osteoarthritis. Osteoarthritis Cartilage. 2009;17(9):1236–43.
McDougall JJ, Schuelert N, Bowyer J. Cathepsin K inhibition reduces CTXII levels and joint pain in the guinea pig model of spontaneous osteoarthritis. Osteoarthritis Cartilage. 2010;18(10):1355–7.
Chappard D, Libouban H, Mindeholm L, Basle MF, Legrand E, Audran M. The cathepsin K inhibitor AAE581 induces morphological changes in osteoclasts of treated patients. Microsc Res Tech. 2010;73(7):726–32.
Malfait AM. Modelling pain in post-traumatic osteoarthritis of the knee. Pain. 2012;153(2):257–8.
Malfait AM, Ritchie J, Gil AS, Austin JS, Hartke J, Qin W, et al. ADAMTS-5 deficient mice do not develop mechanical allodynia associated with osteoarthritis following medial meniscal destabilization. Osteoarthritis Cartilage. 2010;18(4):572–80.
McCulloch DR, Le Goff C, Bhatt S, Dixon LJ, Sandy JD, Apte SS. Adamts5, the gene encoding a proteoglycan-degrading metalloprotease, is expressed by specific cell lineages during mouse embryonic development and in adult tissues. Gene Expr Patterns. 2009;9(5):314–23.
Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4(8):617–29.
Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10(11):1361–8.
Malfait AM, Seymour AB, Gao F, Tortorella MD, Le Graverand-Gastineau MP, Wood LS, et al. A role for PACE4 in osteoarthritis pain: evidence from human genetic association and null mutant phenotype. Ann Rheum Dis. 2012;71(6):1042–8.
Malfait AM, Arner EC, Song RH, Alston JT, Markosyan S, Staten N, et al. Proprotein convertase activation of aggrecanases in cartilage in situ. Arch Biochem Biophys. 2008;478(1):43–51.
Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, et al. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest. 1996;97(3):761–8.
Song RH, Tortorella MD, Malfait AM, Alston JT, Yang Z, Arner EC, et al. Aggrecan degradation in human articular cartilage explants is mediated by both ADAMTS-4 and ADAMTS-5. Arthritis Rheum. 2007;56(2):575–85.
Heinegard D. Proteoglycans and more—from molecules to biology. Int J Exp Pathol. 2009;90(6):575–86.
Wang P, Tortorella M, England K, Malfait AM, Thomas G, Arner EC, et al. Proprotein convertase furin interacts with and cleaves pro-ADAMTS4 (Aggrecanase-1) in the trans-Golgi network. J Biol Chem. 2004;279(15):15434–40.
Tortorella MD, Arner EC, Hills R, Gormley J, Fok K, Pegg L, et al. ADAMTS-4 (aggrecanase-1): N-terminal activation mechanisms. Arch Biochem Biophys. 2005;444(1):34–44.
Longpre JM, McCulloch DR, Koo BH, Alexander JP, Apte SS, Leduc R. Characterization of proADAMTS5 processing by proprotein convertases. Int J Biochem Cell Biol. 2009;41(5):1116–26.
Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G. Biochemical characterization of human collagenase-3. J Biol Chem. 1996;271(3):1544–50.
Kashiwagi M, Tortorella M, Nagase H, Brew K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J Biol Chem. 2001;276(16):12501–4.
Kafienah W, Bromme D, Buttle DJ, Croucher LJ, Hollander AP. Human cathepsin K cleaves native type I and II collagens at the N-terminal end of the triple helix. Biochem J. 1998;331(Pt 3):727–32.
Hou WS, Li Z, Buttner FH, Bartnik E, Bromme D. Cleavage site specificity of cathepsin K toward cartilage proteoglycans and protease complex formation. Biol Chem. 2003;384(6):891–7.
Zack MD, Malfait AM, Skepner AP, Yates MP, Griggs DW, Hall T, et al. ADAM-8 isolated from human osteoarthritic chondrocytes cleaves fibronectin at Ala(271). Arthritis Rheum. 2009;60(9):2704–13.
Grau S, Richards PJ, Kerr B, Hughes C, Caterson B, Williams AS, et al. The role of human HtrA1 in arthritic disease. J Biol Chem. 2006;281(10):6124–9.
Xie DL, Hui F, Meyers R, Homandberg GA. Cartilage chondrolysis by fibronectin fragments is associated with release of several proteinases: stromelysin plays a major role in chondrolysis. Arch Biochem Biophys. 1994;311(2):205–12.
Homandberg GA, Wen C, Hui F. Cartilage damaging activities of fibronectin fragments derived from cartilage and synovial fluid. Osteoarthritis Cartilage. 1998;6(4):231–44.
Stanton H, Ung L, Fosang AJ. The 45 kDa collagen-binding fragment of fibronectin induces matrix metalloproteinase-13 synthesis by chondrocytes and aggrecan degradation by aggrecanases. Biochem J. 2002;364(Pt 1):181–90.
Tsuchiya A, Yano M, Tocharus J, Kojima H, Fukumoto M, Kawaichi M, et al. Expression of mouse HtrA1 serine protease in normal bone and cartilage and its upregulation in joint cartilage damaged by experimental arthritis. Bone. 2005;37(3):323–36.
Chamberland A, Wang E, Jones AR, Collins-Racie LA, LaVallie ER, Huang Y, et al. Identification of a novel HtrA1-susceptible cleavage site in human aggrecan: evidence for the involvement of HtrA1 in aggrecan proteolysis in vivo. J Biol Chem. 2009;284(40):27352–9.
Tortorella M, Tomasselli A, Song L, TenBrink R, Anglin C, Malfait AM. Healthy minds, healthy joints: Defining a novel role for BACE1 in cartilage erosion. OARSI annual meeting; Brussels, Belgium: Osteoarthritis Cartilage; 2010. p. S120.
Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19(2):271–3.
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201.
Henrich S, Cameron A, Bourenkov GP, Kiefersauer R, Huber R, Lindberg I, et al. The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat Struct Biol. 2003;10(7):520–6.
Hall T, Shieh HS, Day JE, Caspers N, Chrencik JE, Williams JM, et al. Structure of human ADAM-8 catalytic domain complexed with batimastat. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68(Pt 6):616–21.
Li Z, Kienetz M, Cherney MM, James MN, Bromme D. The crystal and molecular structures of a cathepsin K:chondroitin sulfate complex. J Mol Biol. 2008;383(1):78–91.
Mosyak L, Georgiadis K, Shane T, Svenson K, Hebert T, McDonagh T, et al. Crystal structures of the two major aggrecan degrading enzymes, ADAMTS4 and ADAMTS5. Protein Sci. 2008;17(1):16–21.
Shieh HS, Mathis KJ, Williams JM, Hills RL, Wiese JF, Benson TE, et al. High resolution crystal structure of the catalytic domain of ADAMTS-5 (aggrecanase-2). J Biol Chem. 2008;283(3):1501–7.
DeLano WL. The PyMOL Molecular Graphics System. 1.3r1 ed. New York: Schrodinger, LLC; 2010.
Acknowledgments
Rachel E. Miller is supported by an Arthritis Foundation Post-Doctoral Fellowship. Anne-Marie Malfait is supported by grant R01AR060364 from the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases. The funding agencies had no part in the preparation of this manuscript.
The articles cited in this review were selected from the authors’ personal libraries of articles and from PubMed searches using the keywords “osteoarthritis”, “proteases”, “inhibitors”, and “animal models”. Selections were made on the basis of the expert opinions of the authors. Searches were performed through December 2012.
Compliance with Ethics Guidelines
ᅟ
Conflict of interest
Anne-Marie Malfait was previously employed by Pfizer. Micky D. Tortorella was previously employed by Pfizer. Anne-Marie Malfait is an associate editor of Osteoarthritis and Cartilage (Elsevier). Rachel E. Miller declares that she has no conflict of interest. Yongzhi Lu declares that he has no conflict of interest.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Osteoarthritis
Rights and permissions
About this article
Cite this article
Miller, R.E., Lu, Y., Tortorella, M.D. et al. Genetically Engineered Mouse Models Reveal the Importance of Proteases as Osteoarthritis Drug Targets. Curr Rheumatol Rep 15, 350 (2013). https://doi.org/10.1007/s11926-013-0350-2
Published:
DOI: https://doi.org/10.1007/s11926-013-0350-2