
Abstract
Pain is a major clinical problem of osteoarthritis (OA). Recently, OA has been thought to be a disease of the whole joint with both destruction of cartilage and inflammatory components such as synovitis and bone marrow lesions. Clinical studies have documented a significant inflammatory soft tissue contribution to the severity and frequency of OA pain. Both clinical and experimental studies have provided evidence for the sensitization of pain pathways during OA, involving pronounced changes in joint nociceptors and changes of the nociceptive processing in the spinal cord, brainstem, and thalamocortical system. Additionally, evidence has been provided for neuropathic pain components in OA models. Concerning molecular mechanisms of OA pain and potential options for pain therapy, studies on nerve growth factor, cytokines, sodium channel blockers, hyaluronic acid preparations, and others are addressed in this review.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) is one of the most frequent causes of chronic pain [1, 2]. Currently, this disease cannot be cured. Therefore, pain therapy is an important medical need that is not met in many cases. Still, many patients undergo surgical joint replacement after many years of pain. Thus, it is urgent to improve our knowledge of pain generation in OA, with the hope of finding better treatment strategies. However, there are several problems with research on OA pain. First, it is not entirely clear which are the most important pathological elements of OA (see below). Second, the origin and the nature of OA pain are a matter of discussion. Third, it is not clear which experimental models are most adequate for investigating the putative mechanisms of human OA pain.
For a long time, the focus of OA research was mainly on the hyaline articular cartilage, which is subjected to progressive and accumulative wear and tear during lifetime. It became apparent, however, that bone remodeling and attrition occur relatively early in the disease, and therefore, OA is now considered a disease of the whole joint [2]. Which factors drive the development of OA is a matter of discussion. While a major driver of progression is aberrant loading (mechanopathology) [2], many authors have stressed the significance of inflammation and enzymatic cartilage degradation in the pathogenesis of OA [3]. Pathologically, OA is characterized by degradation of type II collagen and cartilage destruction, the hallmark of OA [4], synovitis [2–5], and bone marrow lesions [2, 6•], and in the long-term, all tissues of the joint and related structures are affected [2, 7]. The synovial layer of OA joints shows an increased number of lining cells and a mixed inflammatory infiltrate, mainly macrophages and lymphocytes, but the percentage of macrophages, T cells, and B cells is much lower in OA than in rheumatoid arthritis [3, 5]. Neutrophils are absent in synovial fluid, and there is no systemic manifestation of inflammation [4]. The etiology of synovitis is unknown [3]. While both mechanopathology and inflammation play a role in OA, there are certainly different views on the relative importance of these factors.
TNF-α, IL-1ß, other cytokines, chemokines, and metalloproteinases seem to be involved in inflammation and destruction in OA [4, 5]. The OA joint shows an aberrant expression of inflammation-related genes, catabolic genes, nitric oxide (NO), metalloproteinases (MMP-1, MMP-3, MMP-13), cyclooxygenase-2, and cathepsins [4]. These mediators are produced by the synovium and chondrocytes, which respond to biomechanical perturbation by upregulation of the production of proinflammatory cytokines, and stress-induced intracellular signals such as reactive oxygen species (ROS), even in the absence of overt inflammation [4].
Pain in Osteoarthritis
Research into the pain mechanisms of OA is relatively new. Only recent clinical studies have addressed the question of the source and nature of OA pain. Experimental pain research on OA models was also started only in the last few years.
Recent Clinical Research on the Pattern of OA Pain
Intially, pain in OA joints occurs episodically during movements and loading of the joint (hyperalgesia), and this pain may be evoked by specific activities [2]. These are typical features of nociceptive pain. At later stages, constant resting pain may occur, even at night [2, 7]. Thus, there seems to be a gradual increase of pain with OA progress. However, at which time point in the OA process the pain starts is unclear. At advanced states, depressive symptoms and sleep disorders may occur [2]. Furthermore, OA pain may occur together with pain at other locations and neurological disturbances [8].
Which of the pathologies actually cause pain? It is unlikely that the cartilage destruction itself gives rise to pain, because cartilage is normally not innervated. There is some indication that pain mainly arises from secondary synovitis and bone marrow lesions [2, 3]. Pain in knee OA fluctuates with changes of bone marrow lesions and synovitis. When bone marrow lesions become smaller, the pain is reduced, and the risk of frequent pain decreases. By contrast, worsening of synovitis and effusions are associated with increased risk of frequent and more severe pain [6•].
Although there is often a poor relationship between radiographic images and pain, knee pain occurred in a higher proportion of OA patients with Kellgren/Lawrence (K/L) grade 4 than of OA patients with K/L grades 2 and 3. The presence of pain may actually indicate a higher disease activity. In a longitudinal study, knees with frequent pain displayed greater rates of medial cartilage loss (also after adjustment for radiographic OA stage) [9]. In OA of the interphalangeal joints, patients with erosive OA showed more pain and functional impairment than did patients with nonerosive OA [10]. These data also suggest that pain indicates the disease activity.
Pain Research on Experimental OA Models
Pain researchers most often use the monoiodoacetate (MIA) model, although some pain research is carried out with surgical models. MIA-induced OA develops very rapidly. One day after MIA injection into the joint (MIA inhibits glycolysis and is toxic for chondrocytes), chondrocytes are shrunken and show fragmented pyknotic nuclei, the synovial membrane is expanded by fibrin proteinaceous edema fluid, and the joint is mildly infiltrated by lymphocytes, macrophages, and plasma cells. Some days later, the inflammatory response in synovium subsides, necrotic cartilage collapses, and chondrocytes are lost. Osteoclastic activity is increased, the subchondral bone collapses, and fragmentation of bony trabeculae surrounded by osteoclasts and some replacement by fibrous tissue and newly laid trabecular bone appear (for a review, see [11]). The rapidly developing MIA model is clearly different from slowly developing human OA, and it displays in gene arrays substantial differences from human OA [12]. Still, pain researchers trust that pain mechanisms of OA can be identified in this model.
In MIA-induced OA, animals (usually rats) show persistent mechanical hyperalgesia, as, for example, is evident from the assessment of weight bearing. The initial mechanical hyperalgesia, at least, may be caused by inflammatory processes (e.g., [12, 13]). The cytokines TNF and IL-6 were elevated in the earlier stages [14]. Because a proportion of neurons in the dorsal root ganglia (DRG neurons, the cell bodies of primary sensory neurons) begin to express ATF-3 immunoreactivity (a marker of nerve injury), the development of neuropathic pain component was proposed [13, 14]. More recently, an upregulation of galanin and neuropeptide Y and a downregulation of substance P and CGRP in DRG neurons were found, and this represents a pattern typical for neuropathy [12]. A surgically induced destabilization OA model showed changes of neuropeptides in DRGs that were similar to those in the MIA model [12], thus suggesting a neuropathic component also in other OA models. In addition, spinal microglia is activated in the MIA model, also suggesting a neuropathic component [14], because microglial activation is particularly expressed in neuropathy models. A further argument in favor of a neuropathic component is the gabapentin sensitivity of these MIA rats [13]. Gabapentin is used in the treatment of neuropathy in humans. However, it is unknown whether neuropathy is also a typical feature of human OA, and the presence of a neuropathic component in experimental OA models does not imply that OA pain is, for the most part, neuropathic pain. Evidence for the presence of ongoing pain in the MIA model was also reported [15].
In surgically induced experimental OA, the first signs of hyperalgesia may appear only after a few weeks [16]. Even if OA develops more rapidly, it may be difficult to detect the pain, because sometimes only allodynia is noted, whereas no other pain-related behavioral changes may be observed for several weeks [17].
Neuronal Mechanisms of Joint Pain
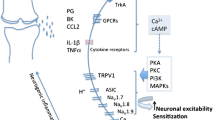
Pain is generated in the nociceptive system. Figure 1 displays a model of the nociceptive system of the joint and indicates major processes that are likely to underlie pain in the OA joint. The following sections will address these mechanisms.

Schema of the nociceptive system of the joint and major neuronal processes at different levels of the neuraxis
Peripheral Neurophysiological Mechanisms
During inflammation, joint nociceptors are substantially sensitized for mechanical stimuli applied to the joint (mechanical sensitization) [18]. Mechanosensitivity of joint nociceptors is also increased in OA models. In the MIA model, the amount of sensitization of joint afferents is correlated with MIA dose [19]. In old guinea pigs with spontaneous OA, joint afferents exhibited increased firing upon noxious movements, but overall, there was no correlation between joint nociception and articular damage [20]. So far, no recordings are available that show a neuropathic activity pattern.
Mechanical sensitization of joint afferents is caused by classical inflammatory mediators, such as prostaglandins, and also by cytokines and other mediators [21], many of which are produced in OA joints (see above). The following paragraphs address mediators and compounds that have been the focus of interest in the last few years and have a relationship to OA.
Nerve Growth Factor
A highly favored target for new drugs against OA pain is nerve growth factor (NGF). In clinical studies, antibodies against NGF produced significant pain relief in moderate to severe OA [22••, 23, 24]. NGF is considered a hot target for several reasons [25]. (1) NGF is an essential growth factor for the development of normal nociceptors. In the adult, a large proportion of nociceptors remain dependent on the trophic effect of NGF, which is required for their structural and functional integrity. These neurons express TrkA receptors (specific receptor for NGF). (2) At inflammatory sites, several cell types produce substantial amounts of NGF, and NGF can directly affect neurons. It enhances currents through TRPV1 channels (cation channels that are opened by heat and capsaicin) and reduces the threshold of thermal excitation. In vivo, the application of NGF generates hyperalgesia, and pretreatment with antibodies to NGF or a fusion protein attached to a modified TrkA receptor attenuates inflammation-induced hypersensitivity. (3) Long-term exposure to NGF increases the expression of TRPV1, bradykinin receptors, purinergic P2X receptors, and Na+ channels and the synthesis of putative nociceptive transmitters such as substance P and CGRP in DRG neurons. (4) NGF stimulates inflammatory cells to release inflammatory compounds. Thus, NGF is considered a key molecule for nociceptor biology.
In mouse OA induced by destabilization of the medial meniscus, NGF was present at day 3 and 16 weeks after the operation, but not in intervals that appeared pain free. A soluble NGF receptor, TrkAd5, effectively suppressed pain in both NGF-positive phases, whereas neutralization of TNF reduced pain only in the first phase, suggesting that OA-associated NGF may not be driven by classical inflammatory mediators [26]. Similarly, another study found in the MIA model a late increase of NGF (in conjunction with a neuropathic component), whereas in the first phase, TNF and IL-6 were elevated [14].
In humans with moderate to severe OA, few injections of a monoclonal antibody against NGF, tenazumab, provided persistent pain relief up to 56 weeks in a dose-dependent fashion and improved function, with a low incidence of side effects [22••]. The initial studies were interrupted because some patients showed during treatment a worsening of OA associated with radiographic evidence of bone necrosis, which required total joint replacements [24]. Recently, however, allowance was given to restart clinical NGF antibody studies.
Cytokines
As was addressed above, cytokines and other mediators are present in human and experimental OA [3–5, 12, 14]. Chondrocyte-produced mediators are not carried away to the systemic circulation but access, via the interstitial tissue fluid and synovial fluid, synovial lining cells and sublining tissues in particular [3]. Concerning pain, some cytokines are now known to act as pro-nociceptive mediators by directly acting on nociceptive neurons that express cytokine receptors. A single injection of either TNF-α [27] or IL-6 [28] into the normal knee joint sensitized Aδ (TNF-α) and C-fibres (TNF-α and IL-6) to innocuous and noxious rotation of the knee joint at a slow time course (taking about 1 h to develop). The so-induced mechanical sensitization persisted for hours, which contrasts to the short-lasting mechanosensitivity increases after intraarterial injection of “classical mediators” (see above). Neutralization of TNF with etanercept prevented the TNF-α-induced mechanical sensitization and partly reversed it once it was established [27]. By contrast, the neutralization of IL-6 (together with its soluble receptor sIL-6R) prevented the generation of mechanical sensitization by IL-6 and attenuated the generation of inflammation-evoked hyperalgesia, but sensitization is difficult to reverse once it is established [28, 29]. Thus, some forms of cytokine-induced mechanical sensitization are quite “stable.” Since many cytokines are present in OA joints, the sensitization of nerve fibers by these cytokines could be an important pain mechanism. Besides TNF-α and IL-6, other cytokines may be important. For example, enhanced expression of IL-1ß gene expression in peripheral blood leukocytes is associated with increased pain and predicts risk for progression of symptomatic knee OA [30]. Whether neutralization of cytokines is an option for treatment of OA pain is an open question.
Sodium Channel Blockers
The opening of voltage-gated sodium channels generates the action potentials (APs) of nerve fibers. However, not all sodium channels are involved in the axonal propagation of APs. The tetrodotoxin-resistant sodium channel Nav1.8, which is almost exclusively expressed in peripheral nociceptors, accounts for most of the inward current of APs of cell bodies in the DRG, and it is thought to be involved in the generation of APs at the sensory endings [21]. The intraarterial injection of A-803467, a blocker of Nav1.8, close to the knee joint, at day 14 of MIA, reduced the firing of joint afferents to noxious rotation (but not of innocuous rotation). Injection of A-803467 into the knee joint reduced the incapacitance and secondary hyperalgesia in MIA rats [31]. Thus, this compound did not block AP propagation but reduced the excitation of the fibers by noxious stimuli. If such sodium channel blockers can be used for pain therapy in the future, OA may be an indication for such a treatment.
Cannabinoids
As an option for pain therapy, cannabinoids were discussed. Cannabinoids can act at CB1 and CB2 receptors. In the rat, CB1 receptor agonists cause antinociception in nociceptors of both the normal knee joint and MIA-induced OA knee joints, but the CB2 receptor agonist GW405833 reduced nociception in the normal knee joint but enhanced mechanonociception in OA knee joints [32]. The enhanced nociception was attributed to an activation of TRPV1 in C fibers, because this sensitizing effect was prevented by a TRPV1 antagonist. Thus, the effect of cannabinoids on mechanonociception in OA joints depends on whether CB1 or CB2 receptors are activated.
Hyaluronic Acid Preparations
For the treatment of OA pain in single big joints, hyaluronic acid (HA) preparations are often injected intraarticularly. The therapeutical benefit of HA was and is being discussed controversially. Comprehensive meta-analyses have stressed the poor quality of many trials and the heterogeneity among the studies and have come to different conclusions, ranging from no effect or a small effect of HA, with highest-molecular-weight HA possibly being more efficacious than lower-molecular-weight HA (for a review, see [33, 34]). Recent studies also have provided conflicting results. One clinical trial used sodium hyaluronate and found no clinical effect beyond placebo effects [35]. Another trial used hylan G-F20 and found a reduction of pain [36]. A further study used a high-molecular-weight HA preparation (GO-ON, 800–1,500 kD) and a lower-molecular-weight HA preparation (sodium hyaluronate, 500–730 kD). Both compounds reduced the pain by about 50 %, but the responder rate was higher after higher-molecular-weight HA (73 % vs. 58 %) [37].
Because the placebo effect is a big confounding factor in patient studies, it is of interest that earlier and recent animal studies showed antinociceptive effects of HA preparations. The intraarticular injection of HA preparations rapidly reduced movement-evoked discharges of rat joint nociceptors [38], thus directly showing an antinociceptive effect, at least for a short time scale. In a rat model of joint pain in which bradykinin and PGE2 were repetitively administered intraarticularly, a single injection of HA preparations at the beginning provided a significant antinociceptive effect (e.g., reduction of hyperalgesia at the knee joint) up to 56 days, in contrast to saline. Cross-linked HA preparations such as Hylan GF20 and NASHA provided a more prolonged antinociceptive effect than did the non-cross-linked sodium hyaluronate [34]. Although these studies did not use OA models, they documented antinociceptive effects of HA preparations.
The mechanisms of action of HA preparations are unclear. HA may provide mechanical protection for the joint due to their viscous properties, or they may cover sensory endings so that these are protected against sensitizing inflammatory mediators [38]. Alternatively, inflammatory mediators might be entrapped in HA through electrostatic interaction. Besides that, HA represents the major component of synovial fluid and fulfills important trophic-metabolic functions [34].
Inhibitors of Angiogenesis
A different and more indirect approach for treating pain may be the inhibition of angiogenesis. In the meniscal transection model of OA in the rat, dexamethasone and indomethacin reduced pain, synovial inflammation, and synovial angiogenesis. Similarly, the angiogenesis inhibitor PPI-2458 reduced pain and angiogenesis. It has been thought that under pathological conditions, nerve fibers, together with vessels, enter newly formed cartilage, and it has been discussed that such an angiogenesis may facilitate inflammation and pain [39].
Central Neurophysiological Mechanisms
Pathological neuronal input from the joint causes complex changes in the central nervous system, which are called central sensitization. In the state of central sensitization, nociceptive neurons at different levels of the neuraxis are hyperexcitable, and hence, the nociceptive processing is amplified. Central sensitization has spinal and supraspinal components.
Spinal Hyperexcitability
In the course of joint inflammation, nociceptive spinal cord neurons with joint input develop a state of hyperexcitability consisting of enhanced responses to mechanical stimulation of the joint (in the innocuous and the noxious range) and lowering of the excitation threshold in high-threshold neurons. Furthermore, the neurons begin to show increased responses to stimuli applied to regions adjacent to and remote from the joint, and the total receptive field can exhibit an enlargement. These changes underlie primary hyperalgesia (at the site of disease) and secondary hyperalgesia (in areas adjacent to and remote from the joint) (for a review, see [18]).
At least at advanced states of OA, patients show signs of central sensitization. They often report widespread pain beyond the OA joint [7] and exhibit lower pressure pain thresholds in cutaneous and subcutaneous structures of the whole leg (for a review, see [11]). Patients with strong local hyperalgesia at the OA knee joint exhibit, in addition, higher pain summation scores upon repetitive stimulation of the OA knee, which is another indicator for central sensitization in severe OA [40]. Experiments on rats with MIA-induced knee OA support these conclusions, because these rats exhibit enhanced spinal responses to mechanical stimulation of the skin at the paw [41, 42].
The mechanisms of central sensitization can be very complex, and the central sensitization during OA, in particular, has not been extensively studied. In OA models (MIA and surgical), the spinal content of substance P and calcitonin gene-related peptide is enhanced [16] (but see [12]). Both neuropeptides are released from sensitized nociceptors and facilitate the generation of spinal hyperexcitability (see [18]). Besides the induction of spinal hyperexcitability by input from the OA joint, the labels of neuropathic pain in the DRGs (e.g., ATF3) raise the possibility that neuropathic mechanisms may contribute as well. Indeed, in the MIA model, the microglia is activated [14], which is typical for neuropathic pain.
Spinal sensitization is often counteracted by local and descending inhibitory mechanisms. Rats with MIA-induced OA exhibited increased levels of the endocannabinoids 2-arachidonoyl glycerol and anandamide. Since spinally applied antagonists at the CB1 and CB2 receptor increased the responses of spinal cord neurons to cutaneous stimulation, it was suggested that endocannabinoids provide tonic spinal inhibition during OA [42].
Because, during OA pain, the whole nociceptive system is sensitized, it should be asked whether the central nervous system should be a target for drug treatment. Opioids suppress the central nociceptive processing, but they are usually not used for the treatment of OA pain. Nonsteroidal anti-inflammatory drugs, which are a major basis of treatment [2], may have central targets. In particular, the selective cyclooxygenase-2 (COX-2) inhibitors were shown to act not only in the periphery, but also in the spinal cord (for a review, see [43]). In a recent study, the spinal application of indomethacin or diclofenac reduced spinal nociception when applied before and during induction of acute inflammation, but it did not reduce the responses of hyperexcitable spinal cord neurons once the hyperexcitability was established. In contrast, two selective COX-2 inhibitors reduced the discharges of neurons also when hyperexcitability was established. However, the latter effect was rather due to an increase of spinal endocannabinoids than to the reduction of spinal prostaglandins [43]. Thus, COX inhibitors differ not only in their side effects.
Descending Pathways Contributing to Pain Generation
Neural pathways descending from the brainstem mediate inhibition and facilitation of nociceptive spinal cord neurons [18]. One form of inhibition—namely, the diffuse inhibitory noxious control (DNIC)—is out of order in patients with severe OA pain and is restored after joint replacement [44]. In addition, descending facilitation may contribute to OA pain. In MIA-induced knee OA, serotoninergic descending facilitation contributes to responses of spinal neurons to innocuous mechanical stimuli applied to the area of cutaneous hyperalgesia in the paw [41]. In hip OA patients, functional imaging showed increased activation of the periaqueductal gray (PAG) in the brain stem during cutaneous stimulation in referred pain areas, which was interpreted as involvement of PAG in central sensitization [45]. These data indicate that the severity of OA pain is partly determined by a distortion of the balance between inhibitory and excitatory modulating descending systems.
Brain Mechanisms of OA Pain
Imaging studies in humans identified brain areas that are involved in the generation of pain. This “cortical pain matrix” is also activated during arthritis [46]. Interestingly, like other chronic pain patients, patients with chronic OA pain exhibit signs of atrophy in the thalamus [47]) and the gray matter of pain-related cortical areas [48]. The functional implication of these changes is not known. Interestingly, these signs of atrophy were reversible after arthroplasty, and thus they seem to be, rather, a consequence than a cause of chronic pain [48].
Conclusions
We begin to get insights into the potential sources and the neuronal mechanisms of OA pain. It remains a challenge, however, to explore in the OA joint, as well as in the nervous system, the sequence of events that cause initial OA pain and lead to progression of OA pain. This also implies defining the nature of OA pain (inflammatory, neuropathic, something else?) at different stages, and such knowledge may provide a rationale for using different types of drugs and nonpharmacological treatments. For example, if there is evidence for substantial central sensitization, a COX inhibitor should be used that has a spinal action upon established spinal sensitization, or if there is evidence for a strong neuropathic component, drugs may be applied that are effective against neuropathic pain.
An intriguing question is whether OA pain will become an indication for a novel approach of pain therapy—namely, the use of biologicals, such as antibodies, against NGF. In the long-term, not only does the benefit of such a therapy have to be shown. It also has to be shown that in spite of the neutralization of the trophic factor NGF, normal nociception is maintained at a sufficient level to provide body protection. Another option for the use of biologicals may be the neutralization of cytokines that play a role in both OA and pain generation. In inflammatory models and in rheumatoid arthritis, neutralization of TNF-α not only reduces the inflammation, but also causes a rapid antinociceptive effect by acting on the nervous system [28, 49].
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Breivik H, Collett B, Ventafridda V, et al. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain. 2006;10:287–333.
Felson DT. Developments in the clinical understanding of osteoarthritis. Arthritis Res Ther. 2009;11:203.
Konttinen YT, Sillat T, Barreto G, et al. Osteoarthritis as an autoinflammatory disease caused by chondrocyte-mediated inflammatory responses. Arthritis Rheum. 2012;64:613–6.
Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol. 2011;23:471–8.
Bondeson J, Blom AB, Wainwright S. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum. 2010;62:647–57.
• Zhang Y, Nevitt M, Niu J, et al. Fluctuation of knee pain and changes in bone marrow lesions, effusions, and synovitis on magnetic resonance imaging. Arthritis Rheum. 2011;63:691–9. This paper reports important data on the sources of pain in patients with knee OA.
Ordeberg G. Evidence of sensitization to pain in human osteoarthritis. In: Felson DT, Schaible H-G, editors. Pain in osteoarthritis. Hoboken: Wiley Blackwell; 2009. p. 199–209.
Hochman JR, Gagliese L, Davis AM, Hawker GA. Neuropathic pain symptoms in a community knee OA cohort. Osteoarthritis Cartilage. 2011;19:647–54.
Eckstein F, Cotofana S, Wirth W, et al. Greater rates of cartilage loss in painful knees than in pain-free knees after adjustment for radiographic disease stage. Arthritis Rheum. 2011;63:2257–67.
Wittoek R, Cruyssen BV, Verbruggen G. Predictors of functional impairment and pain in erosive osteoarthritis of the interphalangeal joints. Arthritis Rheum. 2012;64:1430–6.
Schaible H-G. Joint pain – Basic mechanisms. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack´s textbook of pain. 6th ed. Oxford: Elsevier; 2012. in press.
Im H-J, Kim J-S, Li X, et al. Alteration of sensory neurons and spinal response to an experimental osteoarthritis pain model. Arthritis Rheum. 2010;62:2995–3005.
Ivanavicius SP, Ball AD, Heapy CG, et al. Structural pathology in a rodent model of osteoarthritis is associated with neuropathic pain: increased expression of ATF-3 and pharmacological characterisation. Pain. 2007;128:272–82.
Orita S, Ishikawa T, Miyagi M, et al. Pain-related sensory innervation in monoiodoacetate-induced osteoarthritis in rat knees that gradually develops neuronal injury in addition to inflammatory pain. BMC Musculoskelet Disord. 2011;12:134.
Liu P, Okun A, Guo R-C, et al. Ongoing pain in the MIA model of osteoarthritis. Neurosci Lett. 2011;493:72–5.
Ferland CE, Levrty S, Beaudry F, Vachon P. Gait analysis and pain response of two rodent models of osteoarthritis. Pharmacol Biochem Behav. 2011;97:603–10.
Malfait AM, Ritchie J, Gil AS, et al. ADAMTS-5 deficient mice do not develop mechanical allodynia associated with osteoarthritis following medial meniscal destabilization. Osteoarthritis Cartilage. 2010;18:572–80.
Schaible H-G, Richter F, Ebersberger A, et al. Joint pain. Exp Brain Res. 2009;196:153–62.
Schuelert N, McDougall JJ. Grading of monosodium iodoacetate-induced osteoarthritis reveals a concentration-dependent sensitization of nociception in the knee joint of the rat. Neurosci Lett. 2009;465:184–8.
McDougall JJ, Andruski B, Schuelert N, et al. Unravelling the relationship between age, nociception and joint destruction in naturally occurring osteoarthritis of Dunkin Hartley guinea pigs. Pain. 2009;141:222–32.
Schaible H-G, Ebersberger A, Natura G. Update on peripheral mechanisms of pain: beyond prostaglandins and cytokines. Arthritis Res Ther. 2011;13:210.
•• Lane NE, Schnitzer TJ, Birbara CA, et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med. 2010;363:1521–31. This study shows for the first time the use of a monoclonal antibody to NGF in pain treatment in patients with moderate to severe OA pain.
Nagashima H, Suzuki M, Araki S, et al. Preliminary assessment of the safety and efficacy of tenazumab in Japanese patients with moderate to severe osteoarthritis of the knee: a randomized, double-blind, dose-escalation, placebo-controlled study. Osteoarthritis Cartilage. 2011;19:1405–12.
Schnitzer TJ, Lane NE, Birbara C, et al. Long-term open-label study of tanezumab for moderate to severe osteoarthritis. Osteoarthritis Cartilage. 2011;19:639–46.
Bennett D. NGF, sensitization of nociceptors. In: Schmidt RF, Willis WD, editors. Encyclopedia of pain, volume 2. Berlin, Heidelberg New York, Tokyo: Springer; 2007. p. 1338–42.
McNamee KE, Burleigh A, Gompels LL, et al. Treatment of murine osteoarthritis with TrkAd5 reveals a pivotal role for nerve growth factor in non-inflammatory joint pain. Pain. 2010;149:386–92.
Richter F, Natura G, Loeser S, et al. Tumor necrosis factor-α (TNF-α) causes persistent sensitization of joint nociceptors for mechanical stimuli. Arthritis Rheum. 2010;62:3806–14.
Schaible H-G, Segond von Banchet G, Boettger MK, et al. The role of proinflammatory cytokines in the generation and maintenance of joint pain. Ann N Y Acad Sci. 2010;1193:60–9.
Boettger MK, Leuchtweis J, Kümmel D, et al. Differential effects of locally and systemically administered soluble glycoprotein 130 on pain and inflammation in experimental arthritis. Arthritis Res Ther. 2010;12:R140.
Attur M, Belitskaya-Lévy I, Oh C, et al. Increased interleukin-1ß gene expression in peripheral blood leukocytes is associated with increased pain and predicts risk for progression of symptomatic knee osteoarthritis. Arthritis Rheum. 2011;63:1908–17.
Schuelert N, McDougall JJ. Involvement of Nav1.8 sodium channels in the transduction of mechanical pain in a rodent model of osteoarthritis. Arthritis Res Ther. 2012;14:R5.
Schuelert N, Zhang C, Mogg AJ, et al. Paradoxical effects of the cannabinoid CB(2) receptor agonist GW405833 on rat osteoarthritic knee joint pain. Osteoarthritis Cartilage. 2010;18:1536–43.
Zhang W, Robertson J, Jones AC, Dieppe PA, Doherty M. The placebo effect and its determinants in osteoarthritis: meta-analysis of randomised controlled trials. Ann Rheum Dis. 2008;67:1716–23.
Boettger MK, Kümmel D, Harrison A, Schaible H-G. Evaluation of long-term antinociceptive properties of of stabilized hyaluronic acid preparation (NASHA) in an animal model of repetitive joint pain. Arthritis Res Ther. 2011;13:R110.
Jorgensen A, Stengaard-Pedersen K, Simonsen O, et al. Intra-articular hyaluronan is without clinical effect in knee osteoarthritis: a multicentre, randomised, placebo-controlled, double blind study of 337 patients followed for 1 year. Ann Rheum Dis. 2010;69:1097–102.
Chevalier X, Jerosch J, Goupille P, et al. Single, intraarticular treatment with 6 ml hylan G-F 20 in patients with symptomatic primary osteoarthritis of the knee: a randomised, multicentre, double-blind, placebo controlled trial. Ann Rheum Dis. 2010;69:113–9.
Berenbaum F, Grifka J, Cazzaniga J, et al. A randomised, double-blind, controlled trial comparing two intra-articular hyaluronic acid preparations differing by their molecular weight in symptomatic knee osteoarthritis. Ann Rheum Dis. doi:10.1136/annrheumdis-2011-200972.
Gomis A, Miralles A, Schmidt RF, Belmonte C. Intra-articular injections of hyaluronan solutions of different elastoviscosity reduce nociceptive nerve activity in a model of osteoarthritic knee joint of the guinea pig. Osteoarthritis Cartilage. 2009;17:798–804.
Ashraf S, Mapp PI, Walsh DA. Contributions of angiogenesis to inflammation, joint damage, and pain in a rat model of osteoarthritis. Arthritis Rheum. 2011;63:2700–10.
Arendt-Nielsen L, Nie H, Laursen MB, et al. Sensitization in patients with painful knee osteoarthritis. Pain. 2010;149:573–81.
Rahman W, Bauer CS, Bannister K, et al. Descending serotoninergic facilitation and the antinociceptive effects of pregabalin in a rat model of osteoarthitic pain. Mol Pain. 2009;5:45.
Sagar DR, Staniaszek LE, Okine BN, et al. Tonic modulation of spinal hyperexcitability by the endocannabinoid receptor system in a rat model of osteoarthritis pain. Arthritis Rheum. 2010;62:3666–76.
Telleria-Diaz A, Schmidt M, Kreusch S, et al. Spinal antinociceptive effects of cyclooxygenase inhibition during inflammation: involvement of prostaglandins and endocannabinoids. Pain. 2010;148:26–35.
Kosek E, Ordeberg G. Lack of pressure pain modulation by heterotopic noxious conditioning stimulation in patients with painful osteoarthritis before, but not following surgical pain relief. Pain. 2000;88:69–78.
Gwilym SE, Keltner JR, Warnaby CE, et al. Psychophysical and functional imaging evidence supporting the presence of central sensitization in a cohort of osteoarthritis patients. Arthritis Rheum. 2009;61:1226–34.
Kulkarni B, Bentley DE, Elliott R, et al. Arthritic pain is processed in brain areas concerned with emotions and fear. Arthritis Rheum. 2007;56:1345–54.
Gwilym SE, Filippini N, Douaud G, et al. Thalamic atrophy associated with painful osteoarthritis of the hip is reversible after arthroplasty. Arthritis Rheum. 2010;62:2930–40.
Rodriguez-Raecke R, Niemeier A, Ihle K, et al. Brain gray matter decrease in chronic pain is the consequence and not the cause of pain. J Neurosci. 2009;29:13746–50.
Hess A, Axmann R, Rech J, et al. Blockade of TNF-α rapidly inhibits pain responses in the central nervous system. PNAS. 2011;108:3731–36.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schaible, HG. Mechanisms of Chronic Pain in Osteoarthritis. Curr Rheumatol Rep 14, 549–556 (2012). https://doi.org/10.1007/s11926-012-0279-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-012-0279-x
Keywords
- Bone marrow lesion
- Brain atrophy
- Cannabinoids
- Central sensitization
- COX-2 inhibitor
- Cytokines
- Descending facilitation
- Descending inhibition
- Endocannabinoids
- Hyaluronic acid
- Interleukin-6
- Interleukin-1β
- Joint nociceptor
- Joint pain
- MIA model
- Microglial activation
- Nav1.8
- Nerve growth factor
- Neuropathy
- Osteoarthritis
- Peripheral sensitization
- Sodium channel blocker
- Synovitis
- TNF-α
- TRPV1 channel
- Chronic pain