
Abstract
For the first time in more than 50 years, the US Food and Drug Administration has approved a drug specifically for the treatment of systemic lupus erythematosus (SLE). This drug, belimumab, is a monoclonal antibody that neutralizes the B-cell survival factor, B-lymphocyte stimulator (BLyS). Although belimumab has demonstrated a very favorable safety profile, many SLE patients have failed to clinically improve from belimumab therapy. Three additional BLyS antagonists (atacicept, blisibimod, tabalumab) are currently undergoing clinical testing. These antagonists subtly differ from belimumab in their biologic targets, and each is administered through a route (subcutaneous) that differs from the route through which belimumab is currently delivered (intravenous). Whether these differences will have meaningful consequences for efficacy and safety remains to be determined.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
March 9, 2011, is a day that will be seared in the memories of patients with systemic lupus erythematosus (SLE), their families, and their physicians. On that day, the US Food and Drug Administration (FDA) did something it had not done in more than 50 years—it approved a drug specifically for the treatment of SLE. The drug, belimumab, is a monoclonal antibody (mAb) that binds and neutralizes B-lymphocyte stimulator (BLyS, also commonly known as BAFF). Although the watershed approval of belimumab has instilled renewed hope into the lives of all those touched by SLE, the heterogeneity in clinical response to belimumab clearly highlights the urgent need for further basic and clinical investigation into the BLyS pathway. With that in mind, this review focuses on BLyS and on its closely-related molecule, a proliferation-inducing ligand (APRIL), as well as on the four BLyS antagonists that are already FDA approved (belimumab) or are in clinical development (atacicept, blisibimod, tabalumab).
The BLyS–SLE Connection
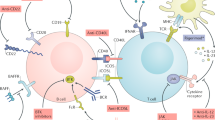
BLyS is a 285-amino acid type-II transmembrane protein member of the tumor necrosis factor ligand superfamily [1, 2]. Cleavage of surface BLyS by a furin protease results in release of a soluble, biologically active 17-kDa molecule [2, 3], which binds to three receptors (BCMA, TACI, and BR3 [BAFFR]) on the surface of B cells (Fig. 1) [4–7]. A combination of in vitro and in vivo studies has demonstrated BLyS to be a vital B-cell survival factor [8–10] and to play important roles in the differentiation of immature B cells to mature B cells [11] and in Ig class switching and Ig production [12]. In contrast, the role for membrane BLyS (as opposed to soluble BLyS) remains uncertain [13].

BLyS engages three receptors (TACI, BCMA, BR3), whereas APRIL engages only two of these (TACI, BCMA). All the BLyS antagonists currently either US Food and Drug Administration approved (belimumab) or undergoing clinical evaluation (atacicept, blisibimod, tabalumab) bind and neutralize BLyS, thereby preventing the binding of BLyS to its receptors. In addition, atacicept also binds and neutralizes APRIL, thereby preventing the binding of APRIL to TACI and BCMA
Non–autoimmune-prone mice that express a BLyS transgene (Tg) and thereby constitutively overexpress BLyS develop SLE-like features, including high titers of circulating anti-dsDNA autoantibodies, immune-complex glomerulonephritis, and proteinuria [14–16]. Moreover, BLyS expression is frequently elevated in human SLE and correlates with disease activity [17–19]. Importantly, SLE-prone NZM 2328 (NZM) mice deficient in BLyS are largely protected from development of clinical disease [20] and are resistant to the SLE-promoting properties of interferon-α [21].
Complicating any elucidation of BLyS biology is the uncertain role of its imperfect doppelganger, APRIL. Although APRIL does not bind to BR3 [6], its three-dimensional structure is sufficiently similar to that of BLyS to permit APRIL to bind to the other two BLyS receptors (BCMA and TACI) (Fig. 1) [22–25]. Moreover, APRIL and BLyS spontaneously form APRIL/BLyS heterotrimers in vivo [26]. What favors formation of heterotrimers rather than BLyS or APRIL homotrimers and whether the potency of the heterotrimers under in vivo conditions is greater than, equal to, or less than those of the BLyS or APRIL homotrimers remain unknown. Of note, APRIL-deficient non-autoimmune mice are either immunologically intact or harbor a modest reduction in serum IgA levels [27, 28], and APRIL-Tg mice do not display signs of autoimmunity [29]. Indeed, the role for APRIL in SLE remains uncertain in large measure due to the absence of any in vitro or in vivo studies in murine or human SLE with agents that selectively block APRIL without also blocking BLyS.
The only reported study to date in SLE that specifically and solely targeted APRIL is a very recent one in which NZM mice were genetically rendered APRIL deficient [30•]. In comparison to NZM wild-type (WT) mice, NZM.April −/− mice harbored increased rather than decreased serum levels of IgG antichromatin antibodies. Moreover, glomerular deposition of IgG and C3 was similar in NZM.April −/− and WT mice; renal histopathology tended to be more severe in NZM.April −/− mice than in WT mice, and development of clinical disease was identical in NZM.April −/− and WT mice. Thus, APRIL is dispensable for development of full-blown SLE in NZM mice and may play a protective role. As deletions present from birth in SLE-prone mice may influence the immune system in a manner that may not be fully applicable to human SLE patients (with intact APRIL genes) receiving therapeutic antagonist agents, the ultimate ramifications of the observations in NZM.April −/− mice for the treatment of human SLE remain to be determined.
Therapeutic Antagonism of BLyS in Murine SLE
In any case, the uncertainty surrounding the role, if any, for APRIL in SLE may have profound consequences for therapeutic agents that target BLyS or its receptors. Treatment of SLE-prone (NZBxNZW)F1 (BWF1) mice or MRL-lpr mice with a BLyS antagonist retards disease progression and improves survival [16, 31]. Of potential concern, the combined antagonism of BLyS and APRIL may be more immunosuppressive than the antagonism of BLyS alone. Indeed, BR3-Ig (which antagonizes BLyS but not APRIL) and TACI-Ig (which antagonizes both BLyS and APRIL) displayed identical clinical efficacy in a head-to-head study despite TACI-Ig having a greater inhibitory effect on humoral immunity than did BR3-Ig [31].
Rationale for Targeting BLyS in Human SLE
For many years, there were no effective B-cell–depleting agents for mice. Accordingly, use of BLyS antagonists as agents that substantially, albeit incompletely, deplete B cells was a logical choice for murine SLE studies. However, very effective B-cell–depleting agents have long been available for humans (eg, rituximab). Thus, one can legitimately ask why the indirect targeting of B cells via a vital survival factor should be preferable to directly targeting the B cells themselves. In the case of BLyS, there are three good reasons.
First, in vivo studies in mice strongly suggest that some (but not all) autoreactive B cells have a greater dependency on BLyS for their survival than do non-autoreactive B cells [32–35]. Antagonism of BLyS could preferentially eliminate pathogenic B cells while sparing those B cells that protect the host from microbial pathogens. This may be especially important from a safety standpoint, as chronic B-cell depletion may be associated with an increased risk of progressive multifocal leukoencephalopathy [36].
Second, a subset of B cells produces interleukin (IL)-10 (B10 cells), and these cells harbor important protective capacities in murine SLE [37•]. Very recently, circulating B10 cells were identified in humans [38•], raising the possibility that these cells play a down-regulatory role not only in murine SLE, but in human SLE as well.
Third, the direct effects of BLyS extend beyond B cells. In mice, BLyS overexpression leads to skewing of in vivo inflammatory responses toward a Th1 cell profile and away from a Th2 cell profile via a B-cell–independent pathway [39]. Moreover, BLyS promotes in vitro generation of Th17 cells, at least in part, through direct effects on T cells [40•]. Thus, therapeutic targeting of BLyS in SLE may not only target pathogenic B cells but may target pathogenic Th1 and/or Th17 cells as well.
Safety and Efficacy of Belimumab
Belimumab is a human IgG1λ mAb that binds and neutralizes soluble BLyS [41]. One of its greatest virtues is its remarkably favorable safety profile. In the phase 1, phase 2, and phase 3 trials of belimumab in SLE, the incidence of adverse events, serious adverse events, and severe adverse events has been essentially identical among belimumab-treated patients as among those who received placebo [42, 43•, 44••, 45••]. That is, patients treated with belimumab at worst did not clinically improve, but, importantly, they were not subjected to toxicities from the medication. Indeed, rates of adverse events and infections over a 6-year period (~1,500 patient-years) have remained stable or have decreased with time [46]. Although this favorable safety profile may, at least in part, be related to the concomitant decrease in corticosteroid usage and/or a preferential loss from long-term cohort studies of patients who are not doing well, the patient retention and lack of long-term morbidity remain striking.
Its outstanding safety profile notwithstanding, actual clinical benefit from belimumab could not be demonstrated until an extensive post-hoc analysis of the failed phase 2 trial results was performed. A novel SLE Response Index (SRI) was used as the measure of clinical response [47••], and the post-hoc analysis revealed efficacy of belimumab (at 52 weeks) to be limited only to those patients who were “seropositive” (serum ANA titer ≥1:80 and/or positive serum anti-dsDNA test) at baseline [43•]. Accordingly, both pivotal phase 3 trials (BLISS-52 and BLISS-76) focused only on such patients. Supporting the tentative conclusion drawn from the post-hoc analysis of the phase 2 trial, patients in the BLISS-52 trial (total n = 865) treated with belimumab (1 or 10 mg/kg) plus standard therapy experienced significantly greater response rates at 52 weeks in comparison to that experienced by patients treated with placebo plus standard therapy [44••]. In addition, patients in the BLISS-76 trial (total n = 819) treated with belimumab (10 mg/kg, but not 1 mg/kg) plus standard therapy also experienced a response rate at 52 weeks greater than that experienced by patients treated with placebo plus standard therapy [45••].
The successful phase 3 trials and the subsequent approval of belimumab by the FDA notwithstanding, there are several caveats that must be stressed.
Frst, more than 40 % of belimumab-treated patients in either trial failed to achieve the primary clinical end point, an SRI response at 52 weeks [44••, 45••]. That is, even among those patients who objectively exhibited B-cell hyperactivity (as assessed by the presence of elevated circulating levels of autoantibodies) at the time of entry into the studies and who, a priori, should have been the most responsive to B-cell–directed therapy, the addition of belimumab to standard therapy did not always improve clinical status. Of note, substantial BLyS-independent autoimmunity and (modest) renal immunopathology also develop over time in BLyS-deficient NZM mice [20]. In addition, global T-cell dysregulation in otherwise non–autoimmune-prone B6 mice leads to rapid development of IgG autoantibodies in a BLyS-independent manner [48]. Furthermore, treatment of BWF1 mice with a BLyS antagonist has, at most, incomplete inhibitory effects on circulating anti-dsDNA autoantibody levels [16, 31]. Thus, the persistence of SLE features among many belimumab-treated human SLE patients may reflect the same BLyS-independent autoimmunity observed in SLE mice.
Second, a significant treatment signal was not appreciated in the BLISS-76 trial at 76 weeks despite a significant treatment signal having been achieved at 52 weeks [45••]. Of note, post-hoc analysis of SRI with higher thresholds of disease activity (change in the SELENA-SLEDAI of 5–10 points rather than only the prescribed 4 points) did demonstrate significant improvement even at 76 weeks among patients treated with the higher belimumab dose. Moreover, the approximately 1,500 patient-year experience to date with belimumab has pointed to durable sustained improvement in SLE disease activity along with a decrease in the frequency of SLE flares [46], suggesting that belimumab may, in fact, have long-term “staying power.” Nonetheless, because much of the accrued experience with belimumab is open label and uncontrolled, the concerns surrounding the duration of effectiveness of belimumab remain incompletely alleviated.
Third, subgroup analysis of the phase 3 results failed to reveal a positive effect for belimumab among black American subjects. Whether this reflects an underpowered cohort of subjects or whether black Americans truly are insensitive to the beneficial effects of belimumab as a consequence of genetic and/or environmental factors is uncertain. Additional studies will be performed to assess efficacy among this population with SLE. Somewhat reassuringly, post-hoc analysis of the 321 seropositive SLE patients in the phase 2 belimumab trial documented the efficacy of belimumab even among black Americans (Human Genome Sciences, unpublished observations). Nevertheless, clinicians will in the meantime have to exercise judgment in evaluating the appropriateness of treating black American SLE patients with belimumab. (Grouping by race is a poor surrogate for identifying disease-susceptibility or treatment-resistance genes. All of us hope that genomics-based studies will soon permit physicians to more accurately predict whether an individual patient will or will not respond to a specific treatment.)
Fourth, patients with “severe active lupus nephritis or central nervous system lupus” were excluded from both BLISS-52 and BLISS-76 [44••, 45••]. Thus, we have no information regarding the efficacy (or safety) of belimumab in those patients who are the sickest and arguably have the greatest unmet therapeutic need. A clinical trial of belimumab in patients with severe active nephritis is being planned, but it will be at least a couple of years before informative results are available.
Fifth, belimumab appears to functionally be a slow-acting drug. That is, even in those patients who did benefit from treatment with belimumab, such benefit was routinely not appreciated until several months after the initiation of belimumab therapy [44••, 45••]. In the BLISS-52 trial, a statistically significant difference in clinical responders between the belimumab 10 mg/kg plus standard therapy and the placebo plus standard therapy groups was realized only by week 24, and it appeared to take even longer in the BLISS-76 trial to realize a statistically significant difference between these two cohorts. It is tempting to speculate that the slow onset of clinical efficacy by belimumab may be related to the persistently increased occupancy by BLyS of BR3 on SLE B cells, even at times when circulating levels of soluble BLyS are not high [49]. For clinical efficacy, belimumab may not only have to neutralize circulating BLyS but may also have overcome the extended tight binding of BLyS to its receptors. Taken together, it is possible that belimumab may not be an ideal agent during the “induction” phase of treatment but may be better suited for “maintenance” therapy.
Other BLyS Antagonists
Atacicept
Although belimumab is the only BLyS antagonist currently approved by the FDA, other BLyS antagonists are at various stages in clinical development (Table 1). The one that has accrued the greatest degree of clinical trials exposure is atacicept, a fusion protein between one of the BLyS receptors (TACI) and the Fc portion of IgG. Atacicept, in contrast to belimumab, binds and neutralizes both BLyS and APRIL (Fig. 1). Neutralization (elimination) of BLyS plus APRIL should have different biological consequences than neutralization (elimination) of BLyS alone. Indeed, in a head-to-head treatment comparison of BR3-Ig (which antagonizes BLyS but not APRIL) with TACI-Ig (which antagonizes both BLyS and APRIL) in SLE mice, treatment with the latter resulted in greater reduction in serum IgM levels, greater reduction in spleen plasma cells (PC), and greater inhibition of IgM responses to a T-cell–dependent antigen than did treatment with the former [31]. Moreover, we recently demonstrated that NZM.Baff −/− .April −/− mice harbor fewer bone marrow PC and have lower serum IgG levels than do NZM.Baff −/− mice [30•]. With regard to clinical outcomes, however, it made no difference whether the SLE mice were treated with BR3-Ig or with TACI-Ig [31], and it made no difference whether the NZM mice were singly deficient in BAFF or doubly deficient in both BAFF and APRIL [30•]. Thus, the net effect of neutralization (elimination) of both BAFF and APRIL may be greater immunosuppression without any readily discernable clinical benefit.
This concern may not be limited to SLE mice but may well extend to human SLE patients. One clinical trial of atacicept (in combination with mycophenolate mofetil) in SLE was prematurely terminated due to an increase in serious infections (ClinicalTrial.gov identifier NCT00573157). Nevertheless, there may still be an important niche for atacicept in the therapeutic armamentarium of the rheumatologist. Even in the phase 1 study of atacicept in SLE, patients treated with drug experienced an approximately 30 % reduction in serum IgG levels by as early as 4 weeks into the trial, whereas patients who received placebo experienced no such reduction [50]. Whereas this rapid decline in serum IgG levels may lie at the heart of the increased incidence of serious infections in the trial that tested atacicept in combination with mycophenolate mofetil, a rapid decline in IgG levels could ultimately prove to be a godsend for patients with severe disease (eg, active nephritis) in whom IgG autoantibodies are a major pathogenic driving force (and for whom there are no data regarding the efficacy/safety of belimumab). Moreover, judicious balancing of concurrent immunosuppressive medications (eg, limiting the dose of mycophenolate mofetil), even in SLE patients with moderate disease activity may largely circumvent any undue proclivity to serious infection while promoting a more rapid clinical response than that seen with belimumab. Overall, the potential for developing serious adverse events related to immunosuppression (eg, serious infections) will likely be greater among patients treated with atacicept than among patients treated with belimumab, but atacicept treatment may turn out to be more beneficial, at least for some patients, if the doses of concurrent immunosuppressive medications can be limited.
Blisibimod
Another BLyS antagonist currently undergoing clinical evaluation is blisibimod (recently known as A-623; originally known as AMG 623). This agent is a “peptibody”—that is, a fusion between the Fc portion of IgG and a peptide sequence selected for its ability to bind with high affinity to BLyS. As the case for belimumab, blisibimod targets only BLyS, but not APRIL (Fig. 1). Accordingly, one might presume that the biological and clinical properties of blisibimod and belimumab should be very similar.
This may not necessarily turn out to be so. Whereas belimumab binds to only soluble BLyS, blisibimod is touted to bind to both soluble and membrane BLyS. (As there are no published papers to date describing the properties and characteristics of blisibimod, one must remain cautious in assuming biological differences.) The hypothesis is that more complete neutralization of BLyS (soluble and membrane) will translate into greater clinical efficacy than that realized by neutralization of only soluble BLyS.
This may not necessarily be the case, as the net in vivo biological activity of membrane BLyS remains unresolved. The phenotype of genetically engineered mice that express membrane BLyS but virtually no soluble BLyS is similar to that of BLyS-deficient mice, with marked reductions in B cells, serum Ig levels, and antigen-specific Ig responses [13]. Accordingly, the clinical response of an SLE patient in whom both membrane and soluble BLyS had been neutralized with blisibimod may be no different from that in a membrane BLyS-replete, but soluble BLyS-deficient SLE patient who had been treated with belimumab. Nonetheless, it would be premature to extrapolate findings made in a murine non-autoimmune host (in whom immunologic tolerance is intact and in whom systemic immune activation and inflammation are quiescent) to a human SLE host (in whom immunologic tolerance has been broken and in whom systemic immune activation and inflammation are ongoing). Thus, it remains to be determined whether joint neutralization of soluble and membrane BLyS is more effective than neutralization of soluble BLyS alone.
Even if only soluble BLyS is biologically active, meaning that blisibimod and belimumab would be directed against the identical biologically active target, the pharmacokinetics of blisibimod administered subcutaneously will still greatly differ from that of belimumab administered intravenously. (Clinical evaluation of subcutaneous belimumab is planned, but from the vantage point of the clinical practitioner, belimumab will remain solely an intravenous drug for at least some years.) On the one hand, because the logistics of administering a subcutaneous medication may be more favorable than those of administering an intravenous medication (ie, home injection vs injection in an infusion center or physician’s office), a subcutaneous medication can be administered more frequently than can an intravenous medication. Thus, the levels of drug can, in principle, be better controlled with the former than with the latter, so the onset of clinically meaningful responses among patients treated with blisibimod may be sooner than in those treated with belimumab. On the other hand, the BLyS-binding region of blisibimod is synthetic, raising the possibility that the host receiving the drug will mount a neutralizing immune response against the drug. In such case, the potency of blisibimod may be severely compromised, and clinical efficacy would suffer.
Tabalumab
The final BLyS antagonist currently undergoing clinical evaluation is tabalumab (originally known as LY2127399), an anti-BLyS mAb that binds to both soluble and membrane BLyS, but not to APRIL (Fig. 1). This mAb has undergone neither phase 1 nor phase 2 evaluation in SLE but is headed directly into two separate phase 3 studies in SLE (ClinicalTrial.gov identifiers NCT01205438 and NCT01196091). Tabalumab is administered subcutaneously, so it may enjoy the same theoretical advantage that blisibimod has in terms of maintaining control of circulating drug levels. Moreover, because tabalumab is not a synthetic agent like blisibimod, tabalumab may be far less immunogenic than blisibimod. Nevertheless, it is difficult to envision how tabalumab would be superior to belimumab (also not synthetic), unless neutralization of membrane BLyS truly adds incremental therapeutic benefit beyond that achieved with neutralization of soluble BLyS alone.
Conclusions
Despite the great excitement generated by BLyS antagonism in murine models of SLE, no unbridled exuberance has been generated from the experience to date with BLyS antagonism in human SLE. Although the approval of belimumab by the FDA for “the treatment of adult patients with active, autoantibody-positive, systemic lupus erythematosus who are receiving standard therapy” unquestionably represents a major milestone in the war against SLE, the true impact of belimumab on SLE will be revealed only as the number of treated patients accrues in the “real world” environment. Belimumab is a step in the right direction, but it realistically is only a small step, with many more steps yet to be taken. Of the three BLyS antagonists currently in clinical testing, the one that has the greatest likelihood of improving on belimumab is atacicept. At the same time, however, atacicept is also the BLyS antagonist with the greatest likelihood of promoting unacceptable toxicities. Whether treatment with atacicept or with any of the other BLyS antagonists leads to a clinical outcome different from that following treatment with belimumab remains to be determined.
Since submission of this manuscript, Huard et al (PLoS ONE 2012;7:e31837) have demonstrated that a monoclonal antibody specific for APRIL can transiently ameliorate clinical disease in BWF1 mice. Whether this amelioration is due to neutralization of APRIL per se or is due to neutralization of APRIL/BLyS heterotrimers and the attendant reduction in BLyS activity was not determined.
References
Papers of particular interest, published recently, have been highlighted as: • of Importance •• of Major Importance
Moore PA, Belvedere O, Orr A, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–3.
Schneider P, MacKay F, Steiner V, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189:1747–56.
Nardelli B, Belvedere O, Roschke V, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204.
Laabi Y, Gras M-P, Brouet J-C, et al. The BCMA gene, preferentially expressed during B lymphoid maturation, is bidirectionally transcribed. Nucleic Acids Res. 1994;22:1147–54.
von Bülow G-U, Bram RJ. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science. 1997;278:138–41.
Thompson JS, Bixler SA, Qian F, et al. BAFF-R, a novel TNF receptor that specifically interacts with BAFF. Science. 2001;293:2108–11.
Yan M, Brady JR, Chan B, et al. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell deficiency. Curr Biol. 2001;11:1547–52.
Thompson JS, Schneider P, Kalled SL, et al. BAFF binds to the tumor necrosis factor receptor-like molecule B cell maturation antigen and is important for maintaining the peripheral B cell population. J Exp Med. 2000;192:129–35.
Do RKG, Hatada E, Lee H, et al. Attenuation of apoptosis underlies B lymphocyte stimulator enhancement of humoral immune response. J Exp Med. 2000;192:953–64.
Batten M, Groom J, Cachero TG, et al. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med. 2000;192:1453–65.
Rolink AG, Tschopp J, Schneider P, Melchers F. BAFF is a survival and maturation factor for mouse B cells. Eur J Immunol. 2002;32:2004–10.
Litinskiy MB, Nardelli B, Hilbert DM, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–9.
Bossen C, Tardivel A, Willen L, et al. Mutation of the BAFF furin cleavage site impairs B-cell homeostasis and antibody response. Eur J Immunol. 2011;41:787–97.
Mackay F, Woodcock SA, Lawton P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–710.
Khare SD, Sarosi I, Xia X-Z, et al. Severe B cell hyperplasia and autoimmune disease in TALL-1 transgenic mice. Proc Natl Acad Sci U S A. 2000;97:3370–5.
Gross JA, Johnston J, Mudri S, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–9.
Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001;44:1313–9.
Zhang J, Roschke V, Baker KP, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001;166:6–10.
Petri M, Stohl W, Chatham W, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008;58:2453–9.
Jacob CO, Pricop L, Putterman C, et al. Paucity of clinical disease despite serological autoimmunity and kidney pathology in lupus-prone New Zealand Mixed 2328 mice deficient in BAFF. J Immunol. 2006;177:2671–80.
Jacob N, Guo S, Mathian A, et al. B cell and BAFF dependence of IFN-α-exaggerated disease in systemic lupus erythematosus-prone NZM 2328 mice. J Immunol. 2011;186:4984–93.
Hahne M, Kataoka T, Schröter M, et al. APRIL, a new ligand of the tumor necrosis factor family, stimulates tumor cell growth. J Exp Med. 1998;188:1185–90.
Marsters SA, Yan M, Pitti RM, et al. Interaction of the TNF homologues BLyS and APRIL with the receptor homologues BCMA and TACI. Curr Biol. 2000;10:785–8.
Yu G, Boone T, Delaney J, et al. APRIL and TALL-1 and receptors BCMA and TACI: system for regulating humoral immunity. Nat Immunol. 2000;1:252–6.
Rennert P, Schneider P, Cachero TG, et al. A soluble form of B cell maturation antigen, a receptor for the tumor necrosis factor family member APRIL, inhibits tumor cell growth. J Exp Med. 2000;192:1677–83.
Roschke V, Sosnovtseva S, Ward CD, et al. BLyS and APRIL form biologically active heterotrimers that are expressed in patients with systemic immune-based rheumatic diseases. J Immunol. 2002;169:4314–21.
Varfolomeev E, Kischkel F, Martin F, et al. APRIL-deficient mice have normal immune system development. Mol Cell Biol. 2004;24:997–1006.
Castigli E, Scott S, Dedeoglu F, et al. Impaired IgA class switching in APRIL-deficient mice. Proc Natl Acad Sci U S A. 2004;101:3903–8.
Stein JV, López-Fraga M, Elustondo FA, et al. APRIL modulates B and T cell immunity. J Clin Invest. 2002;109:1587–98.
• Jacob CO, Guo S, Jacob N, et al. Dispensability of APRIL to the development of systemic lupus erythematosus in NZM 2328 mice. Arthritis Rheum 2012; 64:1610–9. This is the first and only study to date in human or murine SLE to examine the effects of APRIL neutralization/elimination (without concurrent neutralization/elimination of BLyS) on development of SLE features.
Ramanujam M, Wang X, Huang W, et al. Similarities and differences between selective and nonselective BAFF blockade in murine SLE. J Clin Invest. 2006;116:724–34.
Lesley R, Xu Y, Kalled SL, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–53.
Thien M, Phan TG, Gardam S, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–98.
Ota M, Duong BH, Torkamani A, et al. Regulation of the B cell receptor repertoire and self-reactivity by BAFF. J Immunol. 2010;185:4128–36.
Nikbakht N, Migone T-S, Ward CP, Manser T. Cellular competition independent of BAFF/B lymphocyte stimulator results in low frequency of an autoreactive clonotype in mature polyclonal B cell compartments. J Immunol. 2011;187:37–46.
Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;113:4834–40.
• Watanabe R, Ishiura N, Nakashima H, et al. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol 2010;184:4801–4809. This study demonstrated that the B10 subset of B cells has regulatory activity and plays a role in modulating disease activity in murine SLE.
• Iwata Y, Matsushita T, Horikawa M, et al.: Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2011;117:530–541. This study documented the human equivalent of murine B10 regulatory cells. By implication, indiscriminate depletion of B cells would eliminate these regulatory cells and could be counterproductive in the treatment of human SLE.
Sutherland APR, Ng LG, Fletcher CA, et al. BAFF augments certain Th1-associated inflammatory responses. J Immunol. 2005;174:5537–44.
• Zhou X, Xia Z, Lan Q, et al.: BAFF promotes Th17 cells and aggravates experimental autoimmune encephalomyelitis. PLoS ONE 2011;6:e23629. This study documented the ability of BLyS to directly act upon T cells and to promote generation of proinflammatory Th17 cells. The ramification for SLE is that therapeutic neutralization of BLyS may not only be beneficial by inhibiting pathogenic autoreactive B cells, but may also be beneficial by inhibiting generation of pathogenic Th17 cells.
Baker KP, Edwards BM, Main SH, et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum. 2003;48:3253–65.
Furie R, Stohl W, Ginzler EM, et al. Biologic activity and safety of belimumab, a neutralizing anti-B-lymphocyte stimulator (BLyS) monoclonal antibody: a phase I trial in patients with systemic lupus erythematosus. Arthritis Res Ther. 2008;10:R109.
• Wallace DJ, Stohl W, Furie RA, et al.: A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum 2009;61:1168–1178. The post-hoc analysis of this phase 2 study of belimumab in SLE set the stage for the seminal phase 3 studies that ultimately led to approval of belimumab by the FDA.
•• Navarra SV, Guzmán RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–31. The BLISS-52 [44••] and BLISS-76 [45••] trials are the successful phase 3 studies in SLE that led to approval of belimumab by the FDA.
•• Furie R, Petri M, Zamani O, et al.: A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum 2011;63:3918–3930. The BLISS-52 [44••] and BLISS-76 [45••] trials are the successful phase 3 studies in SLE that led to approval of belimumab by the FDA.
Merrill JT, Furie RA, Wallace DJ, et al. Sustained disease improvement and safety profile over the 1500 patient-year experience (6 years) with belimumab in patients with systemic lupus erythematosus (SLE). Arthritis Rheum. 2011;63:S222–3.
•• Furie RA, Petri MA, Wallace DJ, et al.: Novel evidence-based systemic lupus erythematosus responder index. Arthritis Rheum 2009;61:1143–1151. This paper describes the development of the novel SLE Responder Index (SRI), the instrument that was utilized in the BLISS-52 and BLISS-76 trials and that is now being utilized in several other SLE clinical trials.
Stohl W, Jacob N, Quinn III WJ, et al. Global T cell dysregulation in non-autoimmune-prone mice promotes rapid development of BAFF-independent, systemic lupus erythematosus-like autoimmunity. J Immunol. 2008;181:833–41.
Carter RH, Zhao H, Liu X, et al. Expression and occupancy of BAFF-R on B cells in systemic lupus erythematosus. Arthritis Rheum. 2005;52:3943–54.
Dall’Era M, Chakravarty E, Wallace D, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007;56:4142–50.
Disclosure
Dr. Stohl has received clinical trial support from Human Genome Sciences, Eli Lilly and Company, and Amgen.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stohl, W. Biologic Differences Between Various Inhibitors of the BLyS/BAFF Pathway: Should We Expect Differences Between Belimumab and Other Inhibitors in Development?. Curr Rheumatol Rep 14, 303–309 (2012). https://doi.org/10.1007/s11926-012-0254-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-012-0254-6