
Abstract
All cells comprising the skeleton—chondrocytes, osteoblasts, and osteoclasts—contain both the vitamin D receptor and the enzyme CYP27B1 required for producing the active metabolite of vitamin D, 1,25 dihydroxyvitamin D. Direct effects of 25 hydroxyvitamin D and 1,25 dihydroxyvitamin D on these bone cells have been demonstrated. However, the major skeletal manifestations of vitamin D deficiency or mutations in the vitamin D receptor and CYP27B1, namely rickets and osteomalacia, can be corrected by increasing the intestinal absorption of calcium and phosphate, indicating the importance of indirect effects. On the other hand, these dietary manipulations do not reverse defects in osteoblast or osteoclast function that lead to osteopenic bone. This review discusses the relative importance of the direct versus indirect actions of vitamin D on bone, and provides guidelines for the clinical use of vitamin D to prevent/treat bone loss and fractures.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rickets became a public health problem with the movement of the population from the farms to the cities during the Industrial Revolution. Various foods such as cod liver oil and irradiation of other foods including plants were found to prevent or cure this disease, leading eventually to the discovery of the active principle—vitamin D. Given that rickets in children and osteomalacia in adults are the most striking manifestations of vitamin D deficiency, and that the major phenotype in humans and mice lacking a functional vitamin D receptor (VDR) or the enzyme CYP27B1 (25OHD 1α-hydroxylase) producing the active metabolite of vitamin D 1,25(OH)2D, vitamin D signaling is clearly established as critical for bone health. But are its effects direct or indirect? VDRs are found in all cell types of the skeleton: chondrocytes, osteoblasts, osteocytes, and osteoclasts. Likewise, CYP27B1 is also expressed in these cells. Moreover, hundreds of publications have shown direct effects of 1,25(OH)2D and 25OHD on various bone cells making the case for a direct effect. On the other hand, vitamin D deficiency as well as animals (and humans) lacking a functional VDR or CYP27B1, can be successfully treated by increasing the calcium and phosphate content of the diet. Following a brief introduction to the vitamin D endocrine system, I review the data examining the issue of whether the effects of vitamin D on bone are direct or indirect, then conclude with a discussion of current guidelines for the use of vitamin D, not only to prevent rickets and osteomalacia, but the more subtle problems of osteoporosis and fractures.
Overview of Vitamin D Production, Metabolism, and Function
Vitamin D Production, Metabolism to Active Forms, and Serum Transport
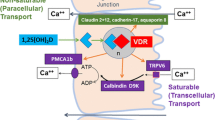
Vitamin D3 is produced in the skin from 7-dehydrocholesterol by ultraviolet (UV) irradiation, which breaks the B ring to form pre-D3. Pre-D3 isomerizes to D3 or with continued UV irradiation to tachysterol and lumisterol [1]. D3 is preferentially removed from the skin, bound to vitamin D binding protein (DBP). Vitamin D is also found in small quantities in the diet, but is a common supplement. This can be in the form of D2 or D3, which differ in their side chains impacting both their affinity for DBP and subsequent metabolism, but which for the sake of this discussion will be treated as equivalent. The liver and other tissues metabolize vitamin D, whether from the skin or oral ingestion, to 25OHD, the principal circulating form of vitamin D, by several enzymes of which CYP27A1 (mitochondrial) and CYP2R1 (microsomal) are the best studied. 25OHD is then further metabolized to 1,25(OH)2D principally in the renal proximal tubule by the enzyme CYP27B1, although other cells such as epidermal keratinocytes, parathyroid gland, intestinal epithelium, macrophages, and various bone cells and chondrocytes contain this enzyme [2•].
1,25(OH)2D is the principal hormonal form of vitamin D, responsible for most of its biologic actions. The production of 1,25(OH)2D in the kidney is tightly controlled, being stimulated by parathyroid hormone (PTH), and inhibited by calcium, phosphate, and fibroblast growth factor 23 (FGF23). Extrarenal production of 1,25(OH)2D as in keratinocytes, macrophages, and osteoblasts is under different control, being stimulated primarily by cytokines such as tumor necrosis factor-α, interferon-γ, and interleukin (IL)-1β [2•, 3]. 1,25(OH)2D reduces 1,25(OH)2D levels in cells by decreasing production or by stimulating its catabolism through the induction of CYP24A1, the 24-hydroxylase [4]. 25OHD and 1,25(OH)2D are hydroxylated in the 24 position by this enzyme to form 24,25(OH)2D and 1,24,25(OH)3D, respectively. This 24-hydroxylation is generally the first step in the catabolism of these active metabolites, although 24,25(OH)2D and 1,24,25(OH)3D have their own biologic activities, and in particular 24,25(OH)2D may be the preferred metabolite regulating resting zone chondrocyte function [5]. CYP24A1 is induced by 1,25(OH)2D, which serves as an important feedback mechanism to avoid vitamin D toxicity. The vitamin D metabolites are transported in blood bound to DBP and albumin. Very little circulates as the free form [6, 7]. The liver produces DBP and albumin, and these proteins may be lost in protein losing enteropathies or the nephrotic syndrome. Thus individuals with liver, intestinal, or renal diseases that result in low levels of these transport proteins may have low total levels of the vitamin D metabolites without being vitamin D deficient as their free concentrations may be normal [7].
Vitamin D Mechanism of Action
VDR is a transcription factor regulating the expression of genes that mediate its biologic activity [8]. VDR is a member of a rather large family of nuclear hormone receptors that includes the receptors for glucocorticoids, mineralocorticoids, sex hormones, thyroid hormone, and vitamin A metabolites or retinoids. VDR is widely distributed, and is not restricted to those tissues considered the classic target tissues of vitamin D. VDR upon binding to 1,25(OH)2D heterodimerizes with other nuclear hormone receptors, in particular the family of retinoid X receptors. This complex then binds to special DNA sequences called vitamin D response elements (VDREs) in the promoters (and other regions) of genes that it regulates. A variety of additional proteins called coactivators bind to the activated VDR/retinoid X receptor (RXR) heterodimers to form a bridge from the VDR/RXR complex to the proteins responsible for transcription such as RNA polymerase II or to help unravel the chromatin at the site of the gene via recruitment of histone acetyl transferases (HATs), allowing transcription to proceed [9]. The vitamin D receptor interacting protein complex (DRIP or Mediator) is an example of the first type of coactivator complex; the steroid receptor coactivator family (SRC 1–3) are examples of the latter type of coactivator. Different genes vary in their regulation by these coactivators. In addition to coactivators there are a number of corepressors. Corepressors typically work by recruiting histone deacetylases to the gene, which reverse the actions of HAT, leading to a reduction in access to the gene by the transcription machinery. Like the coactivators, the corepressors can be specific for different genes, and in either case different cells differentially express these coregulators, providing some specificity for the actions of 1,25(OH)2D and VDR.
In addition to regulating gene expression, 1,25(OH)2D has a number of nongenomic actions including the ability to acutely stimulate various signaling pathways and calcium transport across the plasma membrane. The mechanisms mediating these nongenomic actions and their physiologic significance remain unclear, although both a membrane form of the VDR [10] as well as a distinct membrane protein originally called MARRS (membrane-associated rapid response steroid binding protein) now referred to as PD1A3 (protein disulfide isomerase associated protein 3) [11•, 12] have been implicated. Similarly, it is not clear that all actions of the VDR require the ligand 1,25(OH)2D. The best example of this is the hair loss in animals and patients with certain VDR mutations [13, 14] but not in animals and patients with mutations in CYP27B1 [15, 16]. As mentioned, the VDR is widely distributed, and the actions of 1,25(OH)2D are quite varied. This review focuses on bone.
Vitamin D Effects on Bone
Overview of Bone Formation
Bone develops intramembranously (eg, skull) or from cartilage (endochondral bone formation; eg, long bones with growth plates). Intramembranous bone formation occurs when osteoprogenitor cells proliferate and produce osteoid, a type I collagen-rich matrix. The osteoprogenitor cells differentiate into osteoblasts, which then deposit calcium phosphate crystals into the matrix to produce woven bone. This bone is remodeled into mature lamellar bone. Endochondral bone formation is initiated by the differentiation of mesenchymal stem cells into chondroblasts that produce the proteoglycan-rich type II collagen matrix. These cells continue to differentiate into hypertrophic chondrocytes that shift from making type II collagen to producing type X collagen. These cells also initiate the degradation and calcification of the matrix by secreting matrix vesicles filled with degradative enzymes such as metalloproteinases and phospholipases, alkaline phosphatase (thought to be critical for the mineralization process by hydrolyzing the mineralization inhibitor pyrophosphate), and calcium phosphate crystals. Vascular invasion and osteoclastic resorption are stimulated by the production of vascular endothelial growth factor (VEGF) and other chemotactic factors from the degraded matrix. The hypertrophic chondrocytes also begin to produce markers of osteoblasts such as osteocalcin, osteopontin, and type I collagen resulting in the initial deposition of osteoid. Terminal differentiation of the hypertrophic chondrocytes and the subsequent calcification of the matrix are markedly impaired in vitamin D deficiency leading to the flaring of the ends of the long bones and the rachitic rosary along the costochondral junctions of the ribs, classic features of rickets. Although supply of adequate amounts of calcium and phosphate may correct most of these defects in terminal differentiation and calcification, the vitamin D metabolites, 1,25(OH)2D and 24,25(OH)2D, have been shown to exert distinct roles in the process of endochondral bone formation.
The VDR makes its first appearance in the fetal rat at day 13 of gestation in the condensing mesenchyme of the vertebral column [17], then subsequently in osteoblasts and the proliferating and hypertrophic chondrocytes by day 17 [17]. However, fetal development is quite normal in vitamin D–deficient rats [18] and VDR knockout mice [13], suggesting that vitamin D and the VDR are not critical for skeletal formation. Rickets develops postnatally, becoming most manifest after weaning.
Vitamin D-Regulated Bone Formation
As noted in the introduction nutritional vitamin D deficiency, altered vitamin D responsiveness such as VDR mutations (hereditary vitamin D-resistant rickets), and deficient production of 1,25(OH)2D consequent to CYP27B1 mutations (pseudo-vitamin D deficiency) all have rickets as their main phenotype. This indicates that vitamin D, and in particular 1,25(OH)2D, is of critical importance to bone. Furthermore, both VDR and CYP27B1 are found in bone cells [19, 20], as is CYP27B1 [3, 21–24, 25•, 26•]. However, the rickets resulting from vitamin D deficiency or VDR mutations (or knockouts) can be corrected by supplying adequate amounts of calcium and phosphate by infusions or orally [27–33], or in the case of the VDR knockout by expressing the VDR solely in the intestine [34•]. This would suggest that vitamin D metabolites are unimportant for bone, or that substantial redundancy has been built into the system. I prefer the latter explanation given that numerous examples exist demonstrating the close interaction between calcium and vitamin D with respect to their compensatory/synergistic actions, and bone is no exception [35•].
A further complicating factor in determining the role of vitamin D metabolites in bone is the multitude of effects these metabolites have on systemic calcium homeostatic mechanisms, which themselves impact on bone. The lack of vitamin D results in hypocalcemia and hypophosphatemia, which as implied above is sufficient to cause rickets. Moreover, part of the skeletal phenotype in vitamin D deficiency is also due to the hyperparathyroidism that develops in the vitamin D-deficient state as PTH has its own actions on bone and cartilage. Furthermore, vitamin D metabolites can alter the responsiveness of bone to growth hormone [36], and the expression and/or secretion of a large number of skeletally derived factors including insulin-like growth factor-1 [37], its receptor [38], and binding proteins [39, 40], transforming growth factor-β [41], VEGF [42], IL-6 [43], IL-4 [44], and endothelin receptors [45], all of which can exert effects on bone of their own as well as modulate the actions of the vitamin D metabolites on bone.
Understanding the impact of vitamin D metabolites on bone is additionally complicated by species differences, differences in responsiveness of bone and cartilage cells according to their states of differentiation, and differences in responsiveness in terms of the vitamin D metabolite being examined. For example, 1,25(OH)2D added in the early stages of osteoblast cultures inhibited the expression of collagen 1 and alkaline phosphatase but stimulated their expression when added to more differentiated cultures [46]. Similarly, it has been shown that osteoblasts obtained from VDR knockout mice show increased expression of alkaline phosphatase, bone sialoprotein, and osteocalcin, and mineralize more readily than osteoblasts from wild-type mice in vitro [47] and in vivo [48]. However, other studies have shown an increase in osteoblast differentiation markers with 1,25(OH)2D or 25OHD [3, 21, 49], and overexpressing the VDR in mature osteoblasts also leads to increased bone [50, 51]. Osteocalcin and osteopontin in human and rat cells have well-described VDREs in their promoters [52–54], but the mouse does not [55]. Moreover, alkaline phosphatase and the COL1A1 and COL1A2 genes producing type I collagen do not have clearly defined VDREs, so it remains unclear how these genes are regulated by 1,25(OH)2D. These maturation-dependent effects of 1,25(OH)2D on bone cell function may explain the surprising ability of excess 1,25(OH)2D to block mineralization leading to hyperosteoidosis [56, 57], as such doses may prevent the normal maturation of osteoblasts.
Vitamin D-Regulated Chondrocyte Differentiation and Function
The impairment of endochondral bone formation observed in vitamin D deficiency is associated with decreased alkaline phosphatase activity of the hypertrophic chondrocytes [58], alterations in the lipid composition of the matrix [59] perhaps secondary to reduced phospholipase activity [60], and altered proteoglycan degradation [61] due to changes in metalloproteinase activity [61]. Both 1,25(OH)2D and 24,25(OH)2D appear to be required for optimal endochondral bone formation [62]. However, in the CYP24A1 knockout mouse, which fails to produce any 24-hydroxylated metabolites of vitamin D, the skeletal lesion is defective mineralization of intramembranous (not endochondral) bone [63]. Furthermore, the skeletal abnormality appears to be due to high circulating 1,25(OH)2D levels in that crossing this mouse with one lacking the VDR corrects the problem [63]. Whether this reflects species differences between mice and other species (most studies demonstrating the role of 24,25(OH)2D in bone and cartilage have used rats and chicks) remains unknown. Chondrocytes from the resting zone of the growth plate of rats tend to be more responsive to 24,25(OH)2D than 1,25(OH)2D, whereas the reverse is true for chondrocytes from the growth zone with respect to stimulation of alkaline phosphatase activity [64], regulation of phospholipase A2 (stimulation by 1,25(OH)2D, inhibition by 24,25(OH)2D) [65], changes in membrane fluidity (increased by 1,25(OH)2D, decreased by 24,25(OH)2D) [66], and stimulation of protein kinase C activity [67]. These actions of 1,25(OH)2D and 24,25(OH)2D do not require the VDR and are nongenomic in that they take place with isolated matrix vesicles and membrane preparations from these cells [64]. Evidence for a specific receptor for 24,25(OH)2D in chondrocytes is gaining strength (St. Arnaud, personal communication), and the existence of a functional membrane receptor for 1,25(OH)2D in these cells is supported by the observation that an antibody against PD1A3 (one of the putative membrane receptors for 1,25(OH)2D) blocks these effects of 1,25(OH)2D on these cells [68].
Vitamin D-Regulated Bone Resorption
In addition to its role in promoting bone formation, 1,25(OH)2D promotes bone resorption by increasing the number and activity of osteoclasts [69]. These effects may be direct in part, in that the osteoclast contains the VDR and CYP27B1 [24, 25•], and 25OHD promotes their differentiation in the presence of macrophage colony-stimulating factor (m-CSF) and receptor activator of nuclear factor-κB ligand (RANKL) [25•]. However, these same studies showed an inhibition of their resorptive capacity [25•]. Better established is the stimulation of osteoclastogenesis by 1,25(OH)2D via the osteoblast [70]. Osteoblasts produce membrane-associated RANKL that activates RANK on osteoclasts and their hematopoietic precursors. This cell-to-cell contact in combination with m-CSF also produced by osteoblasts stimulates the differentiation of precursors to osteoclasts, and promotes their activity. 1,25(OH)2D regulates this process by inducing RANKL [71], as does PTH, prostaglandin E2, and IL-11, all of which stimulate osteoclastogenesis. 1,25(OH)2D requires the VDR in osteoblasts for this purpose, although the other hormones and cytokines do not. Osteoblasts from VDR knockout mice fail to support 1,25(OH)2D-induced osteoclastogenesis, whereas osteoclast precursors from VDR knockout mice can be induced by 1,25(OH)2D to form osteoclasts in the presence of osteoblasts from wild-type animals [72], indicating that it is the osteoblast that is the key cell responding to 1,25(OH)2D with respect to osteoclast formation.
Effects on Bone: Direct or Indirect
The degree to which vitamin D directly affects bone versus its indirect actions via 1,25(OH)2D stimulation of intestinal calcium and phosphorus absorption remains a matter of debate, although both are clearly involved. VDR-ablated mice (VDR knockout mice) develop secondary hyperparathyroidism, hypocalcemia, and rickets after weaning [13, 14]; similar changes are seen when CYP27B1 is knocked out [15, 16]. However, as noted previously, when VDR knockout or CYP27B1 knockout mice are fed a rescue diet containing high levels of calcium, phosphorus, and lactose, serum ionized calcium and PTH levels are normalized, and rickets and osteomalacia are prevented [30, 32]. This suggests that a major effect of 1,25(OH)2D3 is the provision of calcium and phosphate to bone from the intestine, rather than a direct action on bone. Furthermore, transgenic expression of VDR in the intestine of VDR knockout mice results in normalization of serum calcium, bone density, and bone volume [34•]. However, a more extensive analysis of the effect of the rescue diet on the skeleton of VDR knockout, CYP27B1 knockout, and CYP27B1/VDR double knockout mice demonstrated that even when hypocalcemia and secondary hyperparathyroidism are prevented by the rescue diet, not all changes in osteoblast number, mineral apposition rate, and bone volume are rescued [73]. In particular, these studies demonstrated that the width of the growth remained increased in the CYP27B1 knockout and double knockout. Furthermore, trabecular bone was markedly osteopenic in all knockout models with decreased mineral apposition rates in both cortical and trabecular bone and reductions in alkaline phosphatase expression, all signifying decreased osteoblast number or activity. This conclusion was confirmed by bone marrow stromal cell cultures from these animals demonstrating reductions in colony-forming units and mineralized nodules despite their ingestion of the rescue diet. Osteoclast numbers in the trabeculae were likewise reduced as was expression of RANKL in extracts of the bones. Other studies have selectively deleted VDR [22] or CYP27B1 [26•] in chondrocytes.
These mice would not be expected to have abnormalities in intestinal calcium and phosphate transport or other metabolite abnormalities accompanying the global knockout of these genes. However, while neither mouse model showed a marked alteration in growth plate development, both showed a decrease in vascular invasion at the chondro-osseous junction with decreased osteoclasts and increased poorly mineralized bone in the primary spongiosa. The chondrocyte-specific VDR null mouse had an increase in serum phosphate and 1,25(OH)2D levels, with decreased FGF23 associated with increased expression of CYP27B1 and the sodium phosphate transporter Npt2a in the kidney [22]. A similar decrease in expression of FGF23 in the bone of chondrocyte-specific CYP27B1 knockout was noted (metabolic consequences were not evaluated). These results indicate that vitamin D signaling in the chondrocyte was clearly affecting endochondral bone formation on the one hand and systemic calcium/phosphate homeostasis via changes in FGF23 production on the other.
So to answer the question as to the relative importance of direct versus indirect effects of vitamin D on bone, we can conclude that both are required, and a deficiency in one can be at least partially compensated by the other.
Clinical Implications
Nutritional Requirements
Up to this point we have been primarily concerned with the animal and cellular studies. These studies including those of transgenic animal models have taught us much concerning the direct and indirect actions of vitamin D on bone. The animal models in particular have demonstrated that although the more flagrant manifestations of rickets/osteomalacia in states of vitamin D deficiency, pseudo-vitamin D deficiency (CYP27B1 mutations), and hereditary vitamin D-resistant rickets (VDR mutations) can be prevented with high calcium/phosphate diets, the more subtle and long-term deficits leading to osteopenia/osteoporosis may not be. While rickets and osteomalacia remain clinically relevant problems, it is the role of vitamin D in the prevention/treatment of osteopenia/osteoporosis that commands most clinical attention at present in developed countries. Serum 25OHD levels provide the most useful surrogate for assessing vitamin D status, as the conversion of vitamin D to 25OHD is less well controlled (ie, primarily substrate dependent) than the subsequent conversion of 25OHD to 1,25(OH)2D. 1,25(OH)2D levels, unlike 25OHD levels, are well maintained until the extremes of vitamin D deficiency because of the secondary hyperparathyroidism, and so do not provide a useful index for assessing vitamin D deficiency at least in the initial stages. Furthermore, as discussed earlier, many tissues including bone express CYP27B1 and may be more dependent on circulating 25OHD than circulating 1,25(OH)2D for their vitamin D requirements [74•].
Historically, vitamin D sufficiency was defined as the level of 25OHD sufficient to prevent rickets in children and osteomalacia in adults. Levels of 25OHD below 5 ng/mL (or 12 nM) are associated with a high prevalence of rickets or osteomalacia. However, there is a growing consensus that these lower limits of normal are too low. Recently an expert panel for the Institute of Medicine recommended that a level of 20 ng/mL (50 nM) was sufficient for 97.5 % of the population with respect to prevention of bone disease and fractures, although up to 50 ng/mL (125 nM) was safe [75•]. For individuals between the ages of 1 to 70 years of age, 600 IU of vitamin D was thought to be sufficient to meet these goals, although up to 4,000 IU of vitamin D was considered safe [75•]. These guidelines at least with respect to the lower recommended levels of vitamin D supplementation are unlikely to correct vitamin D deficiency in individuals with obesity, dark complexions, limited capacity for sunlight exposure, or malabsorption. Moreover, the lower end of these recommendations has been considered too low and the upper end too restrictive by a number of vitamin D experts. In a meta-analysis of several randomized clinical trials, Bischoff-Ferrari et al. [76, 77•] concluded that at least 700 to 800 IU of vitamin D per day were required to achieve the 30 ng/mL (75 nM) level of 25OHD that seemed necessary for fracture prevention. Supplemental calcium may enhance the beneficial actions of vitamin D on bone, but calcitriol and its analogues cannot be recommended because of the higher incidence of hypercalcemia/hypercalciuria [78]. Although most studies have focused on postmenopausal females, the recommendations are relevant to males as well [79]. The benefits of vitamin D on fracture prevention are a combination of increased intestinal calcium absorption [80], increased bone mineral density [81], and reduced risks of falls [82].
Vitamin D Treatment Strategies
Adequate sunlight exposure is the most cost-effective means of obtaining vitamin D. Whole-body exposure to sunlight in the summertime has been calculated to provide the equivalent of 10,000 IU of vitamin D [83]. A 0.5 minimal erythema dose of sunlight (ie, half the dose required to produce a slight reddening of the skin) or UVB radiation to the arms and legs that can be achieved in 5 to 10 min on a bright summer day in light-skinned individuals has been calculated to be the equivalent of 3,000 IU of vitamin D [84]. However, concerns regarding the association between sunlight and skin cancer and/or solar aging of the skin have limited this approach, perhaps to the extreme, although it remains a viable option for those unable or unwilling to benefit from oral supplementation.
Studies have demonstrated that on average for every 100 IU of vitamin D supplementation administered, the 25OHD levels rise by 0.5 to 1 ng/mL [83, 85]. For obese individuals or those with malabsorption (including after bariatric surgery) much higher doses are likely to be required. Unfortified food contains little vitamin D with the exception of wild salmon and other fish products such as cod liver oil. Milk and other fortified beverages typically contain 100 IU/8-oz serving. Vitamin D2 is more rapidly cleared than vitamin D3, so if vitamin D2 is used, it needs to be given at least weekly. Toxicity due to vitamin D supplementation has not been observed at doses less than 10,000 IU per day in several studies [86].
Although the debate over the optimal level of vitamin D is not likely to subside soon, my own recommendation is to maintain a 25OHD level of 30 ng/mL by a combination of sunshine and supplementation, a level that is universally recognized as safe and effective. The amount of vitamin D supplementation required to achieve this level will vary substantially among individuals based on where they live (latitude), time of year, amount of unprotected sun exposure, skin color, body mass index, what they eat, and absorption efficiency so that one-size-fits-all recommendations are inadequate. As in most aspects of medicine, clinical judgment is required.
Conclusions
Understanding the relative contributions of direct and indirect actions of vitamin D on bone is complex. The cells in bone and cartilage contributing to skeletal formation and maintenance have both VDR and CYP27B1, and respond directly to both circulating 25OHD and 1,25(OH)2D (circulating and endogenously produced). However, these cells are also responsive to blood levels of calcium and phosphate, elements required for bone formation. Dietary calcium and phosphate can to some extent compensate for deficient vitamin D signaling, and vitamin D can compensate to some extent for deficiencies in calcium and phosphate. But all are involved. Defining the optimal level of vitamin D to maintain bone health remains under debate. But achieving a level of 25OHD around 30 ng/mL is both safe and effective. Additional research will be necessary to determine whether this is the optimal level.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Holick MF, MacLaughlin JA, Clark MB, et al. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science. 1980;210:203–5.
• Bikle DD. Extra renal synthesis of 1,25 dihydroxyvitamin D and its Health Implications. Clin Rev in Bone and Min Metab. 2009;7:114–25. This review of extrarenal production of 1,25(OH) 2 D provides a good perspective for understanding the importance of the ability of tissues including bone to make their own 1,25(OH) 2 D.
van Driel M, Koedam M, Buurman CJ, et al. Evidence for auto/paracrine actions of vitamin D in bone: 1alpha-hydroxylase expression and activity in human bone cells. Faseb J. 2006;20:2417–9.
Xie Z, Munson SJ, Huang N, et al. The mechanism of 1,25-dihydroxyvitamin D(3) autoregulation in keratinocytes. J Biol Chem. 2002;277:36987–90.
Schwartz Z, Ehland H, Sylvia VL, et al. 1alpha,25-dihydroxyvitamin D(3) and 24R,25-dihydroxyvitamin D(3) modulate growth plate chondrocyte physiology via protein kinase C-dependent phosphorylation of extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase. Endocrinology. 2002;143:2775–86.
Bikle DD, Gee E, Halloran B, et al. Free 1,25-dihydroxyvitamin D levels in serum from normal subjects, pregnant subjects, and subjects with liver disease. J Clin Invest. 1984;74:1966–71.
Bikle DD, Siiteri PK, Ryzen E, et al. Serum protein binding of 1,25-dihydroxyvitamin D: a reevaluation by direct measurement of free metabolite levels. J Clin Endocrinol Metab. 1985;61:969–75.
Pike JW. Genome-wide principles of gene regulation by the vitamin D receptor and its activating ligand. Mol Cell Endocrinol. 2011;347:3–10.
Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71.
Zanello LP, Norman AW. Rapid modulation of osteoblast ion channel responses by 1alpha,25(OH)2-vitamin D3 requires the presence of a functional vitamin D nuclear receptor. Proc Natl Acad Sci U S A. 2004;101:1589–94.
• Chen J, Olivares-Navarrete R, Wang Y, et al. Protein-disulfide isomerase-associated 3 (Pdia3) mediates the membrane response to 1,25-dihydroxyvitamin D3 in osteoblasts. J Biol Chem. 2010;285:37041–50. This study addresses the rapid actions of 1,25(OH) 2 D on bone as mediated by a receptor other than VDR. Both VDR and Pdia3 are likely involved in these rapid, nongenomic actions.
Nemere I, Garbi N, Hammerling GJ, et al. Intestinal cell calcium uptake and the targeted knockout of the 1,25D3-MARRS (membrane-associated, rapid response steroid-binding) receptor/PDIA3/Erp57. J Biol Chem. 2010;285:31859–66.
Li YC, Pirro AE, Amling M, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–5.
Yoshizawa T, Handa Y, Uematsu Y, et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. 1997;16:391–6.
Dardenne O, Prud’homme J, Arabian A, et al. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142:3135–41.
Panda DK, Miao D, Tremblay ML, et al. Targeted ablation of the 25-hydroxyvitamin D 1alpha-hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci U S A. 2001;98:7498–503.
Johnson JA, Grande JP, Roche PC, et al. Ontogeny of the 1,25-dihydroxyvitamin D3 receptor in fetal rat bone. J Bone Miner Res. 1996;11:56–61.
Miller SC, Halloran BP, DeLuca HF, et al. Studies on the role of vitamin D in early skeletal development, mineralization, and growth in rats. Calcif Tissue Int. 1983;35:455–60.
Narbaitz R, Stumpf WE, Sar M, et al. Autoradiographic localization of target cells for 1 alpha, 25-dihydroxyvitamin D3 in bones from fetal rats. Calcif Tissue Int. 1983;35:177–82.
Boivin G, Mesguich P, Pike JW, et al. Ultrastructural immunocytochemical localization of endogenous 1,25-dihydroxyvitamin D3 and its receptors in osteoblasts and osteocytes from neonatal mouse and rat calvaria. Bone Miner. 1987;3:125–36.
Atkins GJ, Anderson PH, Findlay DM, et al. Metabolism of vitamin D3 in human osteoblasts: evidence for autocrine and paracrine activities of 1 alpha,25-dihydroxyvitamin D3. Bone. 2007;40:1517–28.
Masuyama R, Stockmans I, Torrekens S, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116:3150–9.
Anderson PH, O’Loughlin PD, May BK, et al. Modulation of CYP27B1 and CYP24 mRNA expression in bone is independent of circulating 1,25(OH)2D3 levels. Bone. 2005;36:654–62.
Kogawa M, Anderson PH, Findlay DM, et al. The metabolism of 25-(OH)vitamin D3 by osteoclasts and their precursors regulates the differentiation of osteoclasts. J Steroid Biochem Mol Biol. 2010;121:277–80.
• Kogawa M, Findlay DM, Anderson PH, et al. Osteoclastic metabolism of 25(OH)-vitamin D3: a potential mechanism for optimization of bone resorption. Endocrinology. 2010;151:4613–25. This study demonstrated that osteoclast precursors can produce 1,25(OH)2D, and that such production stimulates osteoclast differentiation, but surprisingly reduces their resorptive function at least in an osteoclast cell line (RAW264.7).
• Naja RP, Dardenne O, Arabian A, et al. Chondrocyte-specific modulation of Cyp27b1 expression supports a role for local synthesis of 1,25-dihydroxyvitamin D3 in growth plate development. Endocrinology. 2009;150:4024–32. This study of mice in which CYP27B1 was deleted from chondrocytes demonstrated decreased endochondral bone formation similar to the results from Masuyama et al. [22] in which VDR was specifically deleted in chondrocytes. These are strong arguments for a direct action of vitamin D on bone.
Balsan S, Garabedian M, Larchet M, et al. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest. 1986;77:1661–7.
Underwood JL, DeLuca HF. Vitamin D is not directly necessary for bone growth and mineralization. Am J Physiol. 1984;246:E493–8.
Li YC, Amling M, Pirro AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139:4391–6.
Amling M, Priemel M, Holzmann T, et al. Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology. 1999;140:4982–7.
al-Aqeel A, Ozand P, Sobki S, et al. The combined use of intravenous and oral calcium for the treatment of vitamin D dependent rickets type II (VDDRII). Clin Endocrinol (Oxf). 1993;39:229–37.
Dardenne O, Prud’homme J, Hacking SA, et al. Correction of the abnormal mineral ion homeostasis with a high-calcium, high-phosphorus, high-lactose diet rescues the PDDR phenotype of mice deficient for the 25-hydroxyvitamin D-1alpha-hydroxylase (CYP27B1). Bone. 2003;32:332–40.
Weinstein RS, Underwood JL, Hutson MS, et al. Bone histomorphometry in vitamin D-deficient rats infused with calcium and phosphorus. Am J Physiol. 1984;246:E499–505.
• Xue Y, Fleet JC. Intestinal vitamin D receptor is required for normal calcium and bone metabolism in mice. Gastroenterology. 2009;136:1317–27. e1311–1312. This study provides strong evidence for an indirect effect of vitamin D on bone in that replacing the VDR in the intestine of VDR null mice corrected most of the skeletal abnormalities.
• Bikle DD. Vitamin D: an ancient hormone. Exp Dermatol. 2010;20:7–13. This perspective argues for the dual actions of calcium and vitamin D on most tissues such that lack of either can be at least partially compensated for by the other—a situation relevant to an understanding of the relative roles for indirect and direct actions of vitamin D on bone.
Morales O, Faulds MH, Lindgren UJ, et al. 1Alpha,25-dihydroxyvitamin D3 inhibits GH-induced expression of SOCS-3 and CIS and prolongs growth hormone signaling via the Janus kinase (JAK2)/signal transducers and activators of transcription (STAT5) system in osteoblast-like cells. J Biol Chem. 2002;277:34879–84.
Chenu C, Valentin-Opran A, Chavassieux P, et al. Insulin like growth factor I hormonal regulation by growth hormone and by 1,25(OH)2D3 and activity on human osteoblast-like cells in short- term cultures. Bone. 1990;11:81–6.
Kurose H, Yamaoka K, Okada S, et al. 1,25-Dihydroxyvitamin D3 [1,25-(OH)2D3] increases insulin-like growth factor I (IGF-I) receptors in clonal osteoblastic cells. Study on interaction of IGF-I and 1,25-(OH)2D3. Endocrinology. 1990;126:2088–94.
Scharla SH, Strong DD, Mohan S, et al. 1,25-Dihydroxyvitamin D3 differentially regulates the production of insulin-like growth factor I (IGF-I) and IGF-binding protein-4 in mouse osteoblasts. Endocrinology. 1991;129:3139–46.
Moriwake T, Tanaka H, Kanzaki S, et al. 1,25-Dihydroxyvitamin D3 stimulates the secretion of insulin-like growth factor binding protein 3 (IGFBP-3) by cultured human osteosarcoma cells. Endocrinology. 1992;130:1071–3.
Sato T, Ono T, Tuan RS. 1,25-Dihydroxy vitamin D3 stimulation of TGF-beta expression in chick embryonic calvarial bone. Differentiation. 1993;52:139–50.
Wang DS, Yamazaki K, Nohtomi K, et al. Increase of vascular endothelial growth factor mRNA expression by 1,25-dihydroxyvitamin D3 in human osteoblast-like cells. J Bone Miner Res. 1996;11:472–9.
Lacey DL, Grosso LE, Moser SA, et al. IL-1-induced murine osteoblast IL-6 production is mediated by the type 1 IL-1 receptor and is increased by 1,25 dihydroxyvitamin D3. J Clin Invest. 1993;91:1731–42.
Lacey DL, Erdmann JM, Tan HL, et al. Murine osteoblast interleukin 4 receptor expression: upregulation by 1,25 dihydroxyvitamin D3. J Cell Biochem. 1993;53:122–34.
Nambi P, Wu HL, Lipshutz D, et al. Identification and characterization of endothelin receptors on rat osteoblastic osteosarcoma cells: down-regulation by 1,25-dihydroxy- vitamin D3. Mol Pharmacol. 1995;47:266–71.
Owen TA, Aronow MS, Barone LM, et al. Pleiotropic effects of vitamin D on osteoblast gene expression are related to the proliferative and differentiated state of the bone cell phenotype: dependency upon basal levels of gene expression, duration of exposure, and bone matrix competency in normal rat osteoblast cultures. Endocrinology. 1991;128:1496–504.
Sooy K, Sabbagh Y, Demay MB. Osteoblasts lacking the vitamin D receptor display enhanced osteogenic potential in vitro. J Cell Biochem. 2005;94:81–7.
Tanaka H, Seino Y. Direct action of 1,25-dihydroxyvitamin D on bone: VDRKO bone shows excessive bone formation in normal mineral condition. J Steroid Biochem Mol Biol. 2004;89–90:343–5.
Anderson PH, Atkins GJ, Findlay DM, et al. RNAi-mediated silencing of CYP27B1 abolishes 1,25(OH)2D3 synthesis and reduces osteocalcin and CYP24 mRNA expression in human osteosarcoma (HOS) cells. J Steroid Biochem Mol Biol. 2007;103:601–5.
Baldock PA, Thomas GP, Hodge JM, et al. Vitamin D action and regulation of bone remodeling: suppression of osteoclastogenesis by the mature osteoblast. J Bone Miner Res. 2006;21:1618–26.
Gardiner EM, Baldock PA, Thomas GP, et al. Increased formation and decreased resorption of bone in mice with elevated vitamin D receptor in mature cells of the osteoblastic lineage. Faseb J. 2000;14:1908–16.
Demay MB, Gerardi JM, DeLuca HF, et al. DNA sequences in the rat osteocalcin gene that bind the 1,25-dihydroxyvitamin D3 receptor and confer responsiveness to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1990;87:369–73.
Kerner SA, Scott RA, Pike JW. Sequence elements in the human osteocalcin gene confer basal activation and inducible response to hormonal vitamin D3. Proc Natl Acad Sci U S A. 1989;86:4455–9.
Noda M, Vogel RL, Craig AM, et al. Identification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (SPP-1 or osteopontin) gene expression. Proc Natl Acad Sci U S A. 1990;87:9995–9.
Zhang R, Ducy P, Karsenty G. 1,25-dihydroxyvitamin D3 inhibits Osteocalcin expression in mouse through an indirect mechanism. J Biol Chem. 1997;272:110–6.
Wronski TJ, Halloran BP, Bikle DD, et al. Chronic administration of 1,25-dihydroxyvitamin D3: increased bone but impaired mineralization. Endocrinology. 1986;119:2580–5.
Hock JM, Gunness-Hey M, Poser J, et al. Stimulation of undermineralized matrix formation by 1,25 dihydroxyvitamin D3 in long bones of rats. Calcif Tissue Int. 1986;38:79–86.
Kyeyune-Nyombi E, Lau KH, Baylink DJ, et al. 1,25-Dihydroxyvitamin D3 stimulates both alkaline phosphatase gene transcription and mRNA stability in human bone cells. Arch Biochem Biophys. 1991;291:316–25.
Irving JT, Wuthier RE. Histochemistry and biochemistry of calcification with special reference to the role of lipids. Clin Orthop. 1968;56:237–60.
Howell DS, Marquez JF, Pita JC. The nature of phospholipids in normal and rachitic costochondral plates. Arthritis Rheum. 1965;8:1039–46.
Dean DD, Boyan BD, Muniz OE, et al. Vitamin D metabolites regulate matrix vesicle metalloproteinase content in a cell maturation-dependent manner. Calcif Tissue Int. 1996;59:109–16.
Plachot JJ, Du Bois MB, Halpern S, et al. In vitro action of 1,25-dihydroxycholecalciferol and 24,25-dihydroxycholecalciferol on matrix organization and mineral distribution in rabbit growth plate. Metab Bone Dis Relat Res. 1982;4:135–42.
St-Arnaud R, Arabian A, Travers R, et al. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology. 2000;141:2658–66.
Boyan BD, Schwartz Z, Carnes Jr DL, et al. The effects of vitamin D metabolites on the plasma and matrix vesicle membranes of growth and resting cartilage cells in vitro. Endocrinology. 1988;122:2851–60.
Schwartz Z, Boyan B. The effects of vitamin D metabolites on phospholipase A2 activity of growth zone and resting zone cartilage cells in vitro. Endocrinology. 1988;122:2191–8.
Swain LD, Schwartz Z, Caulfield K, et al. Nongenomic regulation of chondrocyte membrane fluidity by 1,25-(OH)2D3 and 24,25-(OH)2D3 is dependent on cell maturation. Bone. 1993;14:609–17.
Sylvia VL, Schwartz Z, Schuman L, et al. Maturation-dependent regulation of protein kinase C activity by vitamin D3 metabolites in chondrocyte cultures. J Cell Physiol. 1993;157:271–8.
Pedrozo HA, Schwartz Z, Rimes S, et al. Physiological importance of the 1,25(OH)2D3 membrane receptor and evidence for a membrane receptor specific for 24,25(OH)2D3. J Bone Miner Res. 1999;14:856–67.
Suda T, Takahashi N, Abe E. Role of vitamin D in bone resorption. J Cell Biochem. 1992;49:53–8.
Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption—a hypothesis. Calcif Tissue Int. 1981;33:349–51.
Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–602.
Takeda S, Yoshizawa T, Nagai Y, et al. Stimulation of osteoclast formation by 1,25-dihydroxyvitamin D requires its binding to vitamin D receptor (VDR) in osteoblastic cells: studies using VDR knockout mice. Endocrinology. 1999;140:1005–8.
Panda DK, Miao D, Bolivar I, et al. Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem. 2004;279:16754–66.
• Anderson PH, Atkins GJ, Turner AG, et al. Vitamin D metabolism within bone cells: effects on bone structure and strength. Mol Cell Endocrinol. 2011;347:42–7. This review summarizes some of the data supporting a direct action of vitamin D on bone.
• Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53–8. This controversial report sets a recommended floor and ceiling for vitamin D supplementation and optimal serum 25OHD levels.
Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Fracture prevention with vitamin D supplementation: a meta-analysis of randomized controlled trials. Jama. 2005;293:2257–64.
• Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med. 2009;169:551–61. This meta-analysis suggests that the lower limits recommended by the Institute of Medicine report are too low to prevent fractures optimally.
Avenell A, Gillespie WJ, Gillespie LD, et al. Vitamin D and vitamin D analogues for preventing fractures associated with involutional and post-menopausal osteoporosis. Cochrane Database Syst Rev (Online) 2009.CD000227.
Ensrud KE, Taylor BC, Paudel ML, et al. Serum 25-hydroxyvitamin D levels and rate of hip bone loss in older men. J Clin Endocrinol Metab. 2009;94:2773–80.
Heaney RP, Dowell MS, Hale CA, et al. Calcium absorption varies within the reference range for serum 25-hydroxyvitamin D. J Am Coll Nutr. 2003;22:142–6.
Bischoff-Ferrari HA, Kiel DP, Dawson-Hughes B, et al. Dietary calcium and serum 25-hydroxyvitamin D status in relation to BMD among U.S. adults. J Bone Miner Res. 2009;24:935–42.
Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of Vitamin D on falls: a meta-analysis. Jama. 2004;291:1999–2006.
Vieth R. Vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999;69:842–56.
Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–81.
Heaney RP, Davies KM, Chen TC, et al. Human serum 25-hydroxycholecalciferol response to extended oral dosing with cholecalciferol. Am J Clin Nutr. 2003;77:204–10.
Hathcock JN, Shao A, Vieth R, et al. Risk assessment for vitamin D. Am J Clin Nutr. 2007;85:6–18.
Disclosure
Conflicts of interest: D.D. Bikle: has received grant support from the National Institutes of Health (RO1 DK054793, AR055924, and AR050023); and receives loyalties from Lange textbook chapter.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bikle, D.D. Vitamin D and Bone. Curr Osteoporos Rep 10, 151–159 (2012). https://doi.org/10.1007/s11914-012-0098-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-012-0098-z