
Abstract
It is well accepted that aging is one of the most prominent risk factors for the initiation and progression of osteoarthritis. One of the most pronounced age-related changes in chondrocytes is the exhibition of a senescent phenotype, which is the result of several factors including the accumulation of reactive oxygen species and advanced glycation end products. Compared with a normal chondrocyte, senescent chondrocytes exhibit an impaired ability to respond to many mechanical and inflammatory insults to the articular cartilage. Furthermore, protein secretion is altered in aging chondrocytes, demonstrated by a decrease in anabolic activity and increased production of proinflammatory cytokines and matrix-degrading enzymes. Together, these events may make the articular cartilage matrix more susceptible to damage and lead to the onset of osteoarthritis. A better understanding of the mechanisms underlying age-related chondrocyte pathophysiology may be critical for the development of novel therapeutic interventions for progressive joint diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) affects at least 27 million Americans, and is the leading cause of disability in the United States. OA is characterized by the breakdown and loss of articular cartilage, which leads to chronic pain during joint movement. The cause of this disease is not known, but aging is the most influential risk factor for developing OA. Whereas 7.6% of the 18- to 44-year-old age group, and 29.8% of the 45- to 64-year-old age group report doctor-diagnosed arthritis, 50% of persons ages 65 years or older are diagnosed with arthritis [1]. Although aging is generally not viewed as the cause of OA, aging events within articular chondrocytes may predispose the joint to damage when exposed to mechanical loads. This review characterizes age-related changes in the articular chondrocyte, discusses the known molecular mechanisms underlying the effects of chondrocyte aging, and then concludes with how an aged chondrocyte can increase the risk of developing OA.
Age-Related Changes in Articular Chondrocytes
The articular cartilage consists of an extracellular matrix composed primarily of type II collagen and proteoglycans, and one cell type—the chondrocyte. The primary role of the chondrocyte is to maintain cartilage homeostasis, in part through the production of extracellular matrix components. With age, chondrocytes exhibit features consistent with a senescent phenotype, including telomere shortening and increased senescence-associated β-galactosidase activity (Table 1) [2, 3]. These age-related changes impair the ability of chondrocytes to maintain the surrounding extracellular matrix. Accordingly, in aged chondrocytes, synthetic activity decreases and the proteoglycans produced are smaller and more irregular [4, 5].
Chondrocyte synthetic activity is regulated by anabolic growth factors [6]. Aged chondrocytes exhibit a reduced responsiveness to growth factors such as insulin-like growth factor-1 (IGF-1) [7, 8], osteogenic protein-1 (OP-1) or bone morphogenic protein-7 [9], and transforming growth factor-β (TGF-β) [10, 11]. For example, TGF-β stimulates proteoglycan synthesis in young animals, but this effect is impaired in old mice [10, 12]. It is hypothesized that age-related alterations in the TGF-β signaling pathway trigger chondrocytes to leave their normally quiescent state to an autolytic phenotype, leading to degradation of the cartilage extracellular matrix [13].
Factors Affecting Articular Chondrocyte Aging
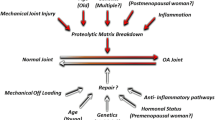
The accumulation of advanced glycation end products (AGEs), increased reactive oxygen species (ROS) production, and age-related changes in joint tissues are several factors that affect chondrocyte aging (Fig. 1). One feature of aging is AGE accumulation in many tissues, including the articular cartilage [14]. AGEs are produced through a nonenzymatic reaction between reducing sugars and free amino groups of proteins, lipids, or nucleic acids [15•]. AGEs are formed within the body, and are also derived from cooking techniques that involve “browning” foods [15•]. Excessive levels of AGEs in the body are pathogenic, and its effects include increased production of oxidative stress and inflammation [16]. In chondrocytes, AGEs increase production of inflammatory cytokine tumor necrosis factor-α (TNF-α) and inflammatory mediators prostaglandin E2 and nitric oxide, suppress collagen II production, and stimulate expression of degradative enzymes matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) [17•, 18, 19]. AGE accumulation also has adverse effects on the cartilage extracellular matrix. AGEs increase collagen crosslinking, which increase tissue stiffness, make cartilage more brittle, and increase susceptibility of cartilage to mechanical failure [20–22]. Although not reported in chondrocytes, AGEs also induce production of ROS in cells such as murine hepatic stellate cells and bone marrow mesenchymal stem cells [23, 24].

Chondrocyte aging and cartilage destruction. Age-related changes within the cartilage extracellular matrix and surrounding joint tissues initiate a cascade of events within the articular chondrocyte that lead to cartilage destruction and susceptibility for the development of osteoarthritis. ADAMTS a disintegrin and metalloproteinase with thrombospondin motifs, MMPs matrix metalloproteinases
ROS play roles in many physiologic processes but have the potential to cause oxidative damage of protein, lipid, and DNA [25]. Mechanisms of oxidative stress suppression include upregulation of antioxidant proteins, such as the ROS-scavenging peroxidases, and enzymes that reverse oxidative damage [26]. However, a loss of reduction-oxidation (redox) homeostasis is linked to degenerative conditions such as Alzheimer’s disease and cancer [27]. Human articular chondrocytes actively produce ROS, and increased levels of ROS were reported in cartilage of old versus young rats [28–31]. Furthermore, the cartilage of old rats exhibited a significant decline in the activity of antioxidant catalase [31]. This redox imbalance may be caused by an age-related decline in the activity and number of mitochondria, which play roles in protecting cells from the harmful effects of ROS [32•]. A consequence of this increase in oxidative stress is DNA damage and telomere shortening, leading to a decline in matrix production, chondrocyte senescence, and apoptosis [33–35]. Increased levels of ROS also upregulate proinflammatory cytokines and MMPs, factors that play a role in the cartilage degradation process [36].
Aging-related changes in joint components also contribute to changes in the chondrocyte. Subchondral bone softening, which occurs during age-related osteoporosis [37], has been predicted to alter the biomechanics of the tibiofemoral joint by increasing maximum tensile strains in the cartilage and magnitudes of joint contact pressure [38]. A decline in quadriceps strength in the elderly population may be another factor responsible for altered joint loading patterns as a consequence of joint laxity [39]. These nonphysiologic loads exerted on the chondrocyte will lead to increased catabolic signaling and cartilage tissue breakdown [40].
Molecular Mechanisms of Chondrocyte Aging
As cellular senescence is a classic feature of chondrocyte aging, several studies have explored mechanisms regulating chondrocyte senescence (Table 1). Shimada et al. used a senescence-accelerated mouse (SAM) to explore the regulation of p21, a molecular marker of senescence [41], by growth arrest and DNA damage-inducible (GADD)45β and CCAAT/enhancer binding protein β (C/EBPβ) [42•]. The GADD45 family of proteins is associated with prosurvival functions in hematopoietic cells [43], and C/EBPβ belongs to a family of transcription factors involved in chondrocyte differentiation [44]. Expectedly, higher expression of senescence-associated β-galactosidase was detected in SAM when compared with controls [42•]. At 58 weeks of age, more chondrocytes in SAM expressed both GADD45β and C/EBPβ. Furthermore, the effect of GADD45β and C/EBPβ on p21 transactivation was determined using a luciferase reporter construct driven by a p21 promoter sequence. The overexpression of GADD45β and C/EBPβ alone slightly increased p21 promoter activity, but the overexpression of both GADD45β and C/EBPβ had a synergistic effect on promoting p21 transcription, suggesting the interactions of GADD45β and C/EBPβ may play important roles in chondrocyte senescence and aging [42•].
Other mediators of cellular senescence include TRF (telomeric repeat binding factor), XRCC5 (x-ray repair complementing defective repair in Chinese hamster cells 5), and SIRT1 (sirtuin 1). TRF1 and TRF2 are telomeric proteins that function to form and maintain telomere structure [45, 46]. XRCC5 is involved in repairing DNA double-strand breaks [47] and SIRT1 is a negative regulator of p53 and prevents growth arrest, senescence, and apoptosis [48]. Oxidative stress in human chondrocytes induced senescence and accelerated telomere shortening [49•]. After acute oxidative insult, TRF1, TRF2, XRCC5, and SIRT1 were upregulated in the early passages of human chondrocytes, but upregulated to a lesser extent in late passages of chondrocytes [49•]. This suggests that TRF proteins, XRCC5, and SIRT1 enable young chondrocytes to cope with oxidative stress by preventing DNA damage accumulation and telomere shortening. Consistently, aged chondrocytes with lower induction levels of these regulatory proteins have a reduced tolerance to oxidative challenge, and the accumulation of DNA damage may trigger chondrocyte senescence.
Membrane protein caveolin-1 is also involved in senescence. Expression of caveolin proteins is increased in the tissues of aged rats [50] and the overexpression of caveolin-1 leads to a senescent phenotype, likely through the p53/p21 pathway [51]. In addition, angiogenic growth factor (AGF) treatment of human chondrocytes downregulated interleukin-1β (IL-1β)–induced caveolin-1 expression and prevented chondrocyte replicative lifespan shortening [52]. Inhibition of p42/p44 mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) abolished the effect of AGF on caveolin-1, suggesting the AGF inhibition of IL-1β–induced chondrocyte aging is mediated, at least in part, by p42/44 MAPK and PI3K [52].
The extent of cell death is unclear in aged populations, but studies suggest there are age-related increases in chondrocyte apoptosis [53, 54], and molecular mechanisms regulating chondrocyte survival have begun to be elucidated (Table 1). IGF-I and OP-1, in addition to their roles in promoting matrix synthesis, contribute to increased survival in normal and OA chondrocytes, suggesting decline of these factors may contribute to age-related cell death [55]. Protein kinase CK2 is a ubiquitously expressed protein with roles in cell growth, proliferation, and apoptosis [56]. Decreased activity of CK2 is reported in chondrocytes of aged rats compared with young controls, and downregulation of CK2 facilitates TNF-α–induced chondrocyte death [57]. Nonhistone chromatin protein high-mobility group box (HMGB) protein 2 is expressed in the superficial zone of mature human chondrocytes, and its expression declines with age. Six-month-old HMGB2−/− mice exhibited reduced cellularity attributable to increased cell death and significant proteoglycan loss, suggesting HMGB2 plays a critical role in the survival of superficial zone chondrocytes [58]. A novel mediator of chondrocyte fate determination might be CBP/p300-interacting transactivator with ED-rich tail 2 (CITED2). Age-related decreases in CITED2 expression have been reported in rat tendon-derived stem/progenitor cells, and were correlated with decreases in cell proliferation and increased cell cycle arrest [59]. CITED2 is expressed in chondrocytes, and has been reported to play a critical role in maintaining cartilage homeostasis through the suppression of MMPs [60, 61]. It will be interesting to investigate whether CITED2 plays a role in cell fate determination in chondrocyte aging.
Chondrocyte Aging and the Development of OA
Aging is not generally viewed as the initiating factor for the development of OA. However, age-related changes within the chondrocyte, including cellular senescence and a reduced responsiveness to growth factors, and external factors affecting chondrocyte aging, such as AGE accumulation and oxidative stress may work in combination to disrupt cartilage homeostasis (Table 1). These changes will make the cartilage matrix more vulnerable to damage and lead to the onset of OA (Fig. 1).
OA is a progressive joint disease that is characterized by cartilage destruction, and affects the structural and functional integrity of the bone and other joint tissues. OA is the most common of all joint diseases, affecting an estimated 15% of the US population [62]. Risk factors for OA include old age, joint trauma, obesity, and heritable genetic factors [63]. The diagnosis of OA is generally performed radiographically and defined by bony changes such as joint space narrowing and osteophyte development [64]. OA commonly affects the knee, hip, and hand joints, and clinical symptoms of OA include joint pain, stiffness, and swelling [65].
The onset of OA is characterized by increased cell proliferation resulting in the formation of chondrocyte clusters and increased synthesis of irregular matrix components including collagens and proteoglycans [66–68]. With OA progression, there is excessive catabolic activity leading to an imbalance of cartilage homeostasis and cartilage matrix breakdown. These catabolic events are mediated largely by proinflammatory cytokines and mediators, MMPs, and ADAMTS [40]. Of note, many characteristics of an aged chondrocyte parallel changes observed in early OA, which might explain why age is highly correlated with the development of OA [69••].
Conclusions
Articular chondrocyte aging is influenced by many systemic and local factors that shift the cell toward a senescent phenotype and initiate catabolic signaling pathways. Together, these factors may contribute to the onset of OA and joint tissue breakdown. Because increased AGE and ROS accumulation are major factors affecting chondrocyte aging, anti-AGE and antioxidant therapies may yield beneficial effects. For example, increased physical fitness is correlated with lower levels of oxidative stress and moderate exercise decreases advanced glycation in small animals, but whether exercise affects levels of AGE and ROS in cartilage is unclear [70, 71]. Therefore, a better understanding of the mechanisms underlying the chondrocyte aging process and its effects on the cartilage extracellular matrix will lead to the development of novel therapeutic strategies to slow or reverse age-related joint degeneration.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Cheng HJ, Hootman JM, Murphy LB, Langmaid GA, Helmick CG. Prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation—United States, 2007–2009. MMWR Morb Mortal Wkly Rep. 2010;59:1261–5.
Martin JA, Buckwalter JA. Aging, articular cartilage chondrocyte senescence and osteoarthritis. Biogerontology. 2002;3:257–64.
Martin JA, Buckwalter JA. The role of chondrocyte senescence in the pathogenesis of osteoarthritis and in limiting cartilage repair. J Bone Joint Surg Am. 2003;85-A Suppl 2:106–10.
Buckwalter JA, Roughley PJ, Rosenberg LC. Age-related changes in cartilage proteoglycans: quantitative electron microscopic studies. Microsc Res Tech. 1994;28:398–408.
Bolton MC, Dudhia J, Bayliss MT. Age-related changes in the synthesis of link protein and aggrecan in human articular cartilage: implications for aggregate stability. Biochem J. 1999;337(Pt 1):77–82.
Fortier LA, Barker JU, Strauss EJ et al. The Role of Growth Factors in Cartilage Repair. Clin Orthop Relat Res 2011.
Loeser RF, Shanker G, Carlson CS, et al. Reduction in the chondrocyte response to insulin-like growth factor 1 in aging and osteoarthritis: studies in a non-human primate model of naturally occurring disease. Arthritis Rheum. 2000;43:2110–20.
Martin JA, Ellerbroek SM, Buckwalter JA. Age-related decline in chondrocyte response to insulin-like growth factor-I: the role of growth factor binding proteins. J Orthop Res. 1997;15:491–8.
Chubinskaya S, Kumar B, Merrihew C, et al. Age-related changes in cartilage endogenous osteogenic protein-1 (OP-1). Biochim Biophys Acta. 2002;1588:126–34.
Blaney Davidson EN, Scharstuhl A, Vitters EL, et al. Reduced transforming growth factor-beta signaling in cartilage of old mice: role in impaired repair capacity. Arthritis Res Ther. 2005;7:R1338–47.
Scharstuhl A, van Beuningen HM, Vitters EL, et al. Loss of transforming growth factor counteraction on interleukin 1 mediated effects in cartilage of old mice. Ann Rheum Dis. 2002;61:1095–8.
van Beuningen HM, van der Kraan PM, Arntz OJ, et al. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab Invest. 1994;71:279–90.
van der Kraan PM, Blaney Davidson EN, van den Berg WB. A role for age-related changes in TGFbeta signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res Ther. 2010;12:201.
Verzijl N, DeGroot J, Thorpe SR, et al. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275:39027–31.
• Uribarri J, Woodruff S, Goodman S, et al. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc. 2010;110:911–16 e12. This is a database of how various food preparation methods affect levels of AGE formation in food.
Ulrich P, Cerami A. Protein glycation, diabetes, and aging. Recent Prog Horm Res. 2001;56:1–21.
• Nah SS, Choi IY, Lee CK, et al. Effects of advanced glycation end products on the expression of COX-2, PGE2 and NO in human osteoarthritic chondrocytes. Rheumatology (Oxford). 2008;47:425–31. This is a study determining the proinflammatory mechanisms of AGEs on chondrocytes.
Nah SS, Choi IY, Yoo B, et al. Advanced glycation end products increases matrix metalloproteinase-1, -3, and −13, and TNF-alpha in human osteoarthritic chondrocytes. FEBS Lett. 2007;581:1928–32.
Huang CY, Lai KY, Hung LF et al. Advanced glycation end products cause collagen II reduction by activating Janus kinase/signal transducer and activator of transcription 3 pathway in porcine chondrocytes. Rheumatology (Oxford) 2011.
Verzijl N, DeGroot J, Ben ZC, et al. Crosslinking by advanced glycation end products increases the stiffness of the collagen network in human articular cartilage: a possible mechanism through which age is a risk factor for osteoarthritis. Arthritis Rheum. 2002;46:114–23.
Chen AC, Temple MM, Ng DM, et al. Induction of advanced glycation end products and alterations of the tensile properties of articular cartilage. Arthritis Rheum. 2002;46:3212–7.
Bank RA, Bayliss MT, Lafeber FP, et al. Ageing and zonal variation in post-translational modification of collagen in normal human articular cartilage. The age-related increase in non-enzymatic glycation affects biomechanical properties of cartilage. Biochem J. 1998;330(Pt 1):345–51.
Guimaraes EL, Empsen C, Geerts A, et al. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol. 2010;52:389–97.
Yang K, Wang XQ, He YS, et al. Advanced glycation end products induce chemokine/cytokine production via activation of p38 pathway and inhibit proliferation and migration of bone marrow mesenchymal stem cells. Cardiovasc Diabetol. 2010;9:66.
Whaley-Connell A, McCullough PA, Sowers JR. The role of oxidative stress in the metabolic syndrome. Rev Cardiovasc Med. 2011;12:21–9.
Avery SV. Molecular targets of oxidative stress. Biochem J. 2011;434:201–10.
Roberts RA, Laskin DL, Smith CV, et al. Nitrative and oxidative stress in toxicology and disease. Toxicol Sci. 2009;112:4–16.
Studer R, Jaffurs D, Stefanovic-Racic M, et al. Nitric oxide in osteoarthritis. Osteoarthritis Cartilage. 1999;7:377–9.
Hiran TS, Moulton PJ, Hancock JT. Detection of superoxide and NADPH oxidase in porcine articular chondrocytes. Free Radic Biol Med. 1997;23:736–43.
Tiku ML, Shah R, Allison GT. Evidence linking chondrocyte lipid peroxidation to cartilage matrix protein degradation. Possible role in cartilage aging and the pathogenesis of osteoarthritis. J Biol Chem. 2000;275:20069–76.
Jallali N, Ridha H, Thrasivoulou C, et al. Vulnerability to ROS-induced cell death in ageing articular cartilage: the role of antioxidant enzyme activity. Osteoarthritis Cartilage. 2005;13:614–22.
• Blanco FJ, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol. 2011;7:161–9. This is a review on the role mitochondrial dysfunction plays in the pathogenesis of OA.
Davies CM, Guilak F, Weinberg JB, et al. Reactive nitrogen and oxygen species in interleukin-1-mediated DNA damage associated with osteoarthritis. Osteoarthritis Cartilage. 2008;16:624–30.
Yudoh K, Nguyen T, Nakamura H, et al. Potential involvement of oxidative stress in cartilage senescence and development of osteoarthritis: oxidative stress induces chondrocyte telomere instability and downregulation of chondrocyte function. Arthritis Res Ther. 2005;7:R380–91.
Nakagawa S, Arai Y, Mazda O, et al. N-acetylcysteine prevents nitric oxide-induced chondrocyte apoptosis and cartilage degeneration in an experimental model of osteoarthritis. J Orthop Res. 2010;28:156–63.
Nelson KK, Melendez JA. Mitochondrial redox control of matrix metalloproteinases. Free Radic Biol Med. 2004;37:768–84.
Marie PJ, Kassem M. Extrinsic mechanisms involved in age-related defective bone formation. J Clin Endocrinol Metab. 2011;96:600–9.
Shirazi R, Shirazi-Adl A. Computational biomechanics of articular cartilage of human knee joint: effect of osteochondral defects. J Biomech. 2009;42:2458–65.
Stevens JE, Binder-Macleod S, Snyder-Mackler L. Characterization of the human quadriceps muscle in active elders. Arch Phys Med Rehabil. 2001;82:973–8.
Sun HB. Mechanical loading, cartilage degradation, and arthritis. Ann N Y Acad Sci. 2010;1211:37–50.
Chang BD, Broude EV, Fang J, et al. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000;19:2165–70.
• Shimada H, Sakakima H, Tsuchimochi K, et al. Senescence of chondrocytes in aging articular cartilage: GADD45beta mediates p21 expression in association with C/EBPbeta in senescence-accelerated mice. Pathol Res Pract. 2011;207:225–31. This is an in vivo model of aging used to elucidate mechanisms of chondrocyte senescence.
Hoffman B, Liebermann DA. Gadd45 modulation of intrinsic and extrinsic stress responses in myeloid cells. J Cell Physiol. 2009;218:26–31.
Martos-Rodriguez A, Santos-Alvarez I, Campo-Ruiz V, et al. Expression of CCAAT/enhancer-binding protein-beta (C/EBPbeta) and CHOP in the murine growth plate. Two possible key modulators of chondrocyte differentiation. J Bone Joint Surg Br. 2003;85:1190–5.
Xin H, Liu D, Songyang Z. The telosome/shelterin complex and its functions. Genome Biol. 2008;9:232.
Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34.
Thacker J, Zdzienicka MZ. The XRCC genes: expanding roles in DNA double-strand break repair. DNA Repair (Amst). 2004;3:1081–90.
Guarente L. Diverse and dynamic functions of the Sir silencing complex. Nat Genet. 1999;23:281–5.
• Brandl A, Hartmann A, Bechmann V, et al. Oxidative stress induces senescence in chondrocytes. J Orthop Res. 2011;29:1114–20. This paper shows molecular mechanisms mediating the effects of oxidative stress on chondrocyte senescence.
Park WY, Park JS, Cho KA, et al. Up-regulation of caveolin attenuates epidermal growth factor signaling in senescent cells. J Biol Chem. 2000;275:20847–52.
Dai SM, Shan ZZ, Nakamura H, et al. Catabolic stress induces features of chondrocyte senescence through overexpression of caveolin 1: possible involvement of caveolin 1-induced down-regulation of articular chondrocytes in the pathogenesis of osteoarthritis. Arthritis Rheum. 2006;54:818–31.
Yudoh K, Shi Y, Karasawa R. Angiogenic growth factors inhibit chondrocyte ageing in osteoarthritis: potential involvement of catabolic stress-induced overexpression of caveolin-1 in cellular ageing. Int J Rheum Dis. 2009;12:90–9.
Aigner T, Hemmel M, Neureiter D, et al. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: a study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 2001;44:1304–12.
Horton Jr WE, Feng L, Adams C. Chondrocyte apoptosis in development, aging and disease. Matrix Biol. 1998;17:107–15.
Loeser RF, Pacione CA, Chubinskaya S. The combination of insulin-like growth factor 1 and osteogenic protein 1 promotes increased survival of and matrix synthesis by normal and osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003;48:2188–96.
Allende JE, Allende CC. Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J. 1995;9:313–23.
Lee SW, Song YS, Lee SY, et al. Downregulation of protein kinase CK2 activity facilitates tumor necrosis factor-alpha-mediated chondrocyte death through apoptosis and autophagy. PLoS One. 2011;6:e19163.
Taniguchi N, Carames B, Ronfani L, et al. Aging-related loss of the chromatin protein HMGB2 in articular cartilage is linked to reduced cellularity and osteoarthritis. Proc Natl Acad Sci U S A. 2009;106:1181–6.
Zhou Z, Akinbiyi T, Xu L, et al. Tendon-derived stem/progenitor cell aging: defective self-renewal and altered fate. Aging Cell. 2010;9:911–5.
Leong DJ, Li YH, Gu XI, et al. Physiological loading of joints prevents cartilage degradation through CITED2. FASEB J. 2011;25:182–91.
Yokota H, Goldring MB, Sun HB. CITED2-mediated regulation of MMP-1 and MMP-13 in human chondrocytes under flow shear. J Biol Chem. 2003;278:47275–80.
Felson DT, Lawrence RC, Dieppe PA, et al. Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med. 2000;133:635–46.
Hinton R, Moody RL, Davis AW, et al. Osteoarthritis: diagnosis and therapeutic considerations. Am Fam Physician. 2002;65:841–8.
Sinkov V, Cymet T. Osteoarthritis: understanding the pathophysiology, genetics, and treatments. J Natl Med Assoc. 2003;95:475–82.
Peat G, Croft P, Hay E. Clinical assessment of the osteoarthritis patient. Best Pract Res Clin Rheumatol. 2001;15:527–44.
Sandell LJ, Aigner T. Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 2001;3:107–13.
Fukui N, Ikeda Y, Ohnuki T, et al. Regional differences in chondrocyte metabolism in osteoarthritis: a detailed analysis by laser capture microdissection. Arthritis Rheum. 2008;58:154–63.
Visco DM, Johnstone B, Hill MA, et al. Immunohistochemical analysis of 3-B-(−) and 7-D-4 epitope expression in canine osteoarthritis. Arthritis Rheum. 1993;36:1718–25.
•• Loeser RF. Age-related changes in the musculoskeletal system and the development of osteoarthritis. Clin Geriatr Med. 2010;26:371–86. This is a review on the effect of aging joint tissues on the development of OA.
Traustadottir T, Davies SS, Su Y, et al. Oxidative stress in older adults: effects of physical fitness. Age (Dordr) 2011.
Boor P, Celec P, Behuliak M, et al. Regular moderate exercise reduces advanced glycation and ameliorates early diabetic nephropathy in obese Zucker rats. Metabolism. 2009;58:1669–77.
Acknowledgment
The author thanks the National Institutes of Health for funding this work (AR52743 and AR47628).
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Leong, D.J., Sun, H.B. Events in Articular Chondrocytes with Aging. Curr Osteoporos Rep 9, 196–201 (2011). https://doi.org/10.1007/s11914-011-0070-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-011-0070-3