
Abstract
Purposeof Review
The purpose of this review is to discuss the current understanding of the pegilodecakin (PEGylated interleukin 10) and its role in the inhibition of tumour growth and metastasis. This review also focuses on clinical data published to date that have evaluated the efficacy and safety of pegilodecakin.
Recent Findings
Pegilodecakin has shown significant promise in preclinical models, notable for decreased tumour burden and fewer sites of metastatic disease across various malignancies. It has been most widely assessed in a phase I/Ib clinical trial against several solid tumours, leading to the phase II and III clinical trials containing pegilodecakin and its combination with other current treatments. However, the updated data have not shown higher efficacy in renal cell carcinoma, metastatic non-small cell lung cancer or pancreatic cancer, with respect to the controls, yet the adverse events presented more mixed results. Further investigation into combination therapies including pegilodecakin is ongoing.
Summary
Pegilodecakin showed promise in preclinical and phase I clinical trials on its efficacy in several solid tumours, with expected regulation of IL-10 signalling pathway observed. However, the phase II and III trials did not justify its application as potential immunotherapy in selected cancers. Further evaluation of pegilodecakin’s efficacy in other cancers, either as monotherapy or in combination with the current treatments, is worth investigating clinically, which warrants to better understand its potential clinical utility.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction to Interleukin 10 and Its Role in Immune System
The molecular mechanism that has been developed in the mammalian immune systems to control the over-activation of their own immune system is known as immunological self-tolerance [1]. In this context, many inhibitory and tightly controlled immune checkpoints are in place to avoid further damage from the innate and/or adaptive immune system to the host. Interleukin-10 (IL-10) is a member of the interleukin-10 family of cytokines and was first thought to have only immunomodulatory activities, contributing to the mechanism of self-tolerance. Indeed, the immunoinhibitory effects of IL-10 were evident through IL-10/IL-10R studies, where not only did the knockout of these genes in mice dramatically increase the chances of developing inflammatory bowel disease, but also enhanced the chances of mice developing colon cancer [2, 3]. Due to the immunoinhibitory activity of IL-10, especially in the tumour microenvironment, blockade of IL-10 receptors using antibodies has been shown to have a therapeutic effect against multiple types of cancer [4•]. Conversely, due to the pleiotropic nature of this cytokine, IL-10 has gained more attention to be used as an immunotherapeutic as it has been shown to possess tumour suppressor effects. The evidence has come from tumour-bearing mice whose cells were gene-engineered to highly express mIL-10, which has been demonstrated to promote tumour rejection [5]. The exact molecular mechanism as to how this occurs is still under debate; nevertheless, it is known that systemic and intratumoural injection of either PEGylated or non-pegylated IL-10 has resulted in the expansion and activation of CD8+ T cells, in addition to higher levels of infiltrating of CD8+ and NK cells in the tumour microenvironment [6,7,8,9, 10•, 11].
Rationale for IL-10 Signalling Regulation
It has been recognised that a better prognosis for cancer patients is correlated with the number of infiltrating and activated CD8+ T cells at the tumour site [12]. Despite the controversial role of IL-10 in tumour immunotherapy due to its known immunosuppressive effects on various hematopoietic cells [4•], early evidence has shown the potential of IL-10 to be tumour-suppressive by promoting CD8+ T cells activation and expansion in vitro [13]. Surprisingly, in addition to the immunostimulatory effects seen in CD8+ T cells, clinical trials of participants with endotoxemia treated with IL-10 have had higher levels of IFN-γ in their serum [14]. These two pieces of evidence have therefore led to the hypothesis that IL-10 could potentially be used for cancer immunotherapy, since IFN-γ upregulates MHC class I/II and favours Th1 response, but not Th17, all of which contribute to better prognosis of cancer patients [7, 9]. Later, the molecular mechanism of how IL-10 was able to promote proliferation and activation of CD8+ T cells was demonstrated by acting on STAT3 and STAT1, with a lesser effect on the latter [15]. Interestingly, the upregulation gene was only observed in tumour infiltrating CD8+ T cells and not in the peripheral lymph [8].
However, although the proof-of-concept that IL-10 could be used as an immunotherapeutic was coming to light, many obstacles must be overcome in order to turn this potential to reach clinical trials. Firstly, although IL-10 had shown promising results by improving the proliferation of tumour CD8+ T cells and activating their immune activities [7], without the need for T-helper cell stimulation [9], there was still no proof of whether it could reach the tumour site through either systemic or subcutaneous application. Secondly, precedents of using other cytokines for treating oncologic diseases, such as TNF-α and IL-12, have failed to reach the market due to their adverse effects in the periphery [16]. In addition, although the concept of IL-10 was being consolidated in combating cancer in vitro and in vivo [5, 6, 9, 11, 17], it was clear that the poor pharmacokinetics of IL-10 largely impedes the drug to become effective for practical and regulatory purposes [9, 10•, 18]. Lastly, IL-10 acting on macrophages and professional antigen-presenting cells has been shown to downregulate their ability to activate and expand T cells [19].
Preclinical Indications for Pegilodecakin
Due to the relatively poor pharmacokinetics of IL-10, most studies designed to comprehend the role of IL-10 in murine models have used engineered IL-10 secreting tumours [6, 9, 18], as the application of this cytokine intravenously or subcutaneously is often rapidly degraded in the liver and cleared by the kidneys [20]. Considering this problem, the formulation of a PEGylated form of IL-10 has been developed. One study has shown that a mono-PEGylation (5 kDa) of IL-10 at its N-terminus increased its t1/2 by 2.7-fold with either subcutaneous or intravenous application, yet the biodistribution of the IL-10 was reduced [21]. In line with this finding, the PEGylation of IL-10 at certain lysine residue(s) using 5 and 20 kDa PEGs increased drug’s retention time in blood after 15 min by around sevenfold, when compared to its non-PEGylated cognate [22]. However, the exact PEGylated lysine residue(s) showing the optimal retention time has not been characterised, due to the rapid degradation of certain PEGylated conformations during the analysis.
Considering this evidence and the reasons previously described for using IL-10 in oncologic diseases, a patent for a mono-PEGylated rIL-10 (pegilodecakin) was filed (Patent number: US7052686B2)[23], which showed promising results in the treatment of multiple malignancies in combination or without other immunotherapeutic drugs, such as immune checkpoint inhibitors, in preclinical settings. Pegilodecakin, refers to a recombinant IL-10 protein with PEG molecules (with a particular range of molecular weight) covalently attached to its N-terminus. Since IL-10 appears as a homodimer, PEGylation can occur at either or both N-termini. The final product is normally a non-homogenous mixture composed of mono- or di-PEGylated forms, where the PEG molecular weight lies between 5 and does not exceed 50 kDa, but usually is within the 5 to 30 kDa range [24]. Most synthetic methods aim to form pegilodecakin with the PEGylation occurred only at one monomer’s N-terminus via a PEG-aldehyde linker, so the attachment is stable, although homogeneous mixture is not achieved due to domain shuffling (Patent number: US10653751B2). The linker reacts with the N-terminus of the proteins, creating imines that are posteriorly reduced using sodium cyanoborohydride, giving the final form of pegilodecakin. Pegilodecakin, using either 12 or 20 kDa of PEG propyladehyde, was demonstrated to have higher inhibition activity after 20 h against all serum inflammatory cytokines measured when compared to its non-PEGylated counterpart (Fig. 1A) [23]. In addition, the activity of higher molecular weight PEG-IL-10 persisted at 72 h when compared to the 12 kDa PEG. Given that pegilodecakin can extend the life span of CD8+ T cells and enhance its cytotoxic activity patients [7], the combination of it with other current treatments in various cancers might offer synergistic effects.

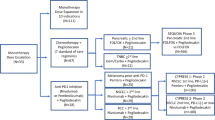
Preclinical and clinical results of pegilodecakin. (A) PEGylated IL-10 with PEG 12 kDa or 20 kDa significantly upregulated TNFα, IL-12p40 and IL-6 post treatment, with respect to non-PEGylated IL-10. The data for graphing was derived from [23]. (B) Pegilodecakin associated treatments in advanced renal cell carcinoma (RCC) of phase Ib clinical trial (NCT02009449). The graph summarising the percentage of patients with different TNM stage at initial diagnosis and the most common reason for discontinuation was constructed based on the results reported in [25•]. AE: adverse events; CD: clinical deterioration; PD: progressive disease. (C) The phase II clinical trial results for pegilodecakin in combination with checkpoint inhibitors in treating metastatic non-small cell lung cancer (NSCLC) [26•]. The control arms received pembrolizumab (CYPRESS 1, NCT03382899) or nivolumab (CYPRESS 2, NCT03382912), while the experimental arms received combination treatments including pegilodecakin. The main outcomes were graphed based on the data presented. ORR: objective response rate; BICR: blinded independent central radiologic review; DCR: disease control rate; mTTR, median time in the therapeutic range; mPFS, progression-free survival; mOS, overall survival. (D) Study schema of the phase III SEQUOIA trial of pegilodecakin in combination with FOLFOX in metastatic pancreatic ductal adenocarcinoma (NCT02923921). This trial compared the efficacy of FOLFOX and FOLFOX + pegilodecakin, including random assignment and patient disposition. FOLFOX, folinic acid, fluorouracil, and oxaliplatin; Gem + Nab Pac, gemcitabine plus nab-paclitaxel; AE, adverse event; PD, progressive disease [27•].
It has been found that mono- and/or di-PEGylation of IL-10 at its N-terminus shows bioactivities, such as increasing the production of IFN-γ in the tumour microenvironment, promoting the expansion and proliferation of cytotoxic T cells via STAT3, and increases MHC formation in macrophages, to multiple other cells from the humoural and adaptive immune system with improved half-life and pharmacokinetics in vivo [8, 9, 28]. RNA-seq analysis of the intravenous application of pegilodecakin has shown elevated expression of IFN-γ only in tumour infiltrating CD8+ T cells, with little to no activity in CD8+ T cells in the secondary lymphoid organ [9]. However, the application of the pegilodecakin did not decrease angiogenesis or infiltrating M1 type macrophages in the tumour microenvironment in Her2 positive breast cancer–bearing mice. Subcutaneous application in mice has shown differences in pegilodecakin uptake depending on whether the cytokine is mono- or di-PEGylated. Overall, although di-PEGylated IL-10 showed slow absorption, PEGylation of both N-termini has shown to remain in blood circulation for longer [21]. Due to the slow absorption and clearance, di-PEGylation is the prevalent isoform circulating drug over time [21]. To date, although the mixture of mono- and di-PEGylated IL-10 produces an effect against multiple hematopoietic cells in vitro and in vivo [29], their magnitudes in term of activating IL-10R remain unclear.
Clinical Implementation of Pegilodecakin
In line with preclinical data demonstrating promising results in the treatment of malignancy [30], pegilodecakin has been submitted to phase I trial (NCT02009449) in treating multiple solid tumours, including renal cell carcinoma (RCC), non-small cell lung cancer (NSCLC), colorectal cancer, melanoma, pancreatic cancer, ovarian cancer, and metastatic castration-resistant prostate cancer, either as a monotherapy or in combination with other current treatments, which is still active and estimated to complete by the end of 2022. Early evidence showed that utilising pegilodecakin improved the outcomes of renal cell carcinoma (RCC) in 4 out of 16 participants [7]. Pegilodecakin upregulated Th1 and Th2 and activated CD8+ T cells without requiring T helper stimulation [7], in addition to elevating Granzyme B and FAS-L in peripheral blood in both NSCLC and PC patients. Interestingly, higher levels of HAL-A, p-STAT3 and LAG3 were also identified in the biopsies of tumours treated with pegilodecakin, all of which are correlated with better cancer prognosis [31, 32]. Thus, phase II and III clinical trials to evaluate the efficacy of pegilodecakin in combination with the standard treatments for NSCLC (pembrolizumab and/or nivolumab) and PC (FOLFOX) were separately initiated.
Clinical Implementation of Pegilodecakin Associated Treatments in Advanced Renal Cell Carcinoma
The phase I clinical trial (NCT02009449) involved pegilodecakin as monotherapy, or in combination with anti-PD-1 inhibitors (pembrolizumab or nivolumab), or tyrosine kinase inhibitor (pazopanib) revealed clinical efficacy in advanced renal cell carcinoma (aRCC) (data cut-off in July 2018) [33•]. The phase Ib dose escalation and expansion study on pegilodecakin in the treatment of 66 patients with advanced metastatic renal cell carcinama (mRCC) was then completed in February 2019 and the final results of cohorts A, G, H and I of IVY phase I study were published [25•, 34•]. A total of 35 patients were allocated to receive pegilodecakin + anti-PD-1 inhibitor (PEG-PD-1) treatment, with respect to 24 and 4 respectively received pegilodecakin monotherapy and pegilodecakin + pazopanib (PEG-PB). The ratios of male to female patients who joined in these treatments were between 2 and 3, and patients with different initial diagnosis of TNM stages were included in the trial (Fig. 1B). Eventually, 58 patients were considered evaluable for further analysis; the ORRs of monotherapy, PEG-PD-1 and PEG-PB were 20%, 43% and 33%, respectively. Tumour burden was reduced notably in the arm treated by PEG-PD-1, which predicted 1-year OS of 76% compared with 50% in the other two arms. In addition, median PFS (mPFS) was found to be 13.9 months in the PEG-PD-1 arm, with respect to 1.8 and 3.7 months in the monotherapy and PEG-PB, respectively. The trial was discontinued, the main reason for discontinuation included progressive disease, clinical deterioration, and adverse events (AEs) (Fig. 1B); anaemia (32%), thrombocytopenia (15%), and hypertriglyceridemia (14%) were among the common Grade 3/4 treatment–related adverse events (TRAEs), which appeared consistent with previous observations [34•, 35]. There were 30 patients who showed serious AEs, with the most frequent events being anaemia (6.1%), dyspnea (6.1%), pyrexia (4.5%) and pneumonia (4.5%). The relatively small sample size is the main limitation of this trial, and the comparison with only anti-PD-1 inhibitors should be considered in further investigation.
Clinical Implementation of Pegilodecakin in Combination with Checkpoint Inhibitors in Metastatic Non-Small Cell Lung Cancer
The phase I IVY trial of pegilodecakin + CPI provided promising efficacy results that led to a randomised phase II trials CYPRESS 1 (N = 101) (NCT03382899) and CYPRESS 2 (N = 52) (NCT03382912) in first-/second-line metastatic Non-Small Cell Lung Cancer (NSCLC), which were cut off in December and August 2019, respectively [26•]. Patients were randomised 1:1 for two arms, and treated with pembrolizumab or pegilodecakin + pembrolizumab on one arm (CYPRESS 1), whereas nivolumab or pegilodecakin + nivolumab was used for the other arm (CYPRESS 2). The key outcomes were summarised in Fig. 1C, showing that the addition of pegilodecakin to either CPI did not improve the efficacy with respect to ORR, PFS, or OS, or mTTR, mPFS, mOS or median duration of therapy. TRAEs ≥ 3 were increased in the experimental arms compared with monotherapy, with the most common AE related to treatment being anaemia, which implied higher toxicity exhibited by the combination treatment and might be associated with a higher rate of discontinuing study treatment. The exploratory post hoc analysis of CYPRESS 1 patients who did not discontinue treatment due to AEs showed ORR (54% versus 44%, 95% CI: 0.6–3.6) and DCR (71% versus 63%, 95% CI: 0.5–3.8), as well as the overlap of the PFS and OS Kaplan–Meier plots. The analysis of cytokines suggested increases in IL-18, Granzyme B, FAS-L, IFN-γ, whereas TGFβ was decreased in the peripheral blood from patients with the treatments including pegilodecakin with respect to the baseline levels, which accorded with the immunostimulatory signals of IL-10R pathway regulated by pegilodecakin. In addition, higher density of CD8+ T cells appeared correlated with higher ORR, DCR, and PFS in both arms of CYPRESS 1.
Clinical Implementation of FOLFOX Combined Pegilodecakin in Metastatic Pancreatic Cancer
Folinic acid, fluorouracil and oxaliplatin (FOLFOX) is a second-line therapy in gemcitabine refractory pancreatic ductal adenocarcinoma (PDAC), which has demonstrated clinical efficacy with acceptable tolerability in patients with pancreatic cancer [36,37,38,39,40]. Besides, the clinical trials of FOLFOX in metastatic colorectal cancer are underway [41,42,43]. Considering that pegilodecakin can enhance the cytotoxicity of CD8 + T cells in vitro and lead to expansion of tumour infiltrating CD8 + T cells in mice, an open-label phase 1b trial was implemented in US (NCT02009449), with data cut-off in February 2019 [44•]. A total of 39 heavily pre-treated patients were subcutaneously treated with pegilodecakin in combination with FOLFOX daily. The most common grade ≥ 3 treatment-emergent adverse events (TEAEs) for the combined treatment included anaemia (17 [43.6%] of 39), thrombocytopenia (21[53.8%] of 39), and neutropenia (13[33.3%] of 39). No substantial immune-related adverse events (irAEs) were observed. The overall response rate (ORR) was 13.6% among the 22 evaluable patients who received the combined treatment, with median time to response of 1.8 months and a median duration of response of 11.5 months. Median PFS (mPFS) and mOS were 2.6 and 6.8 months, respectively. One and 2-year survival rates were estimated to be respectively 36.0% and 24.0%. Notably, two patients exhibited complete responses, and one patient showed 100% tumour reduction.
The compelling results of the phase 1b trial led to a phase III trial (NCT02923921) that comparatively assesses the efficacy of FOLFOX and FOLFOX + pegilodecakin have been implemented in patients with metastatic pancreatic ductal adenocarcinoma (PDAC). A randomised and global phase III trial, namely SEQUOIA, was finalised in 2019[27•] (Fig. 1D). Patients comprised of nearly equal gender from Asia, Europe and North America, took part in this trial, most of them received gemcitabine-containing therapy priorly. It was found that the OS was similar between the arms treated with FOLFOX and FOLFOX + pegilodecakin, which were 5.8 and 6.3 months, respectively (hazard ratio = 1.045; 95% CI, 0.863 to 1.265). The OS rate was estimated to be 19.1% and 14.7% for 1 year in the FOLFOX and FOLFOX + pegilodecakin groups with Kaplan–Meier analysis, respectively. Also, similar levels of progression-free survival (PFS) (2.1 months; HR50.98; 95% CI, 0.81 to 1.19) and ORR (5.6% in FOLFOX and 4.6% in FOLFOX + pegilodecakin) were obtained. Although the safety profile of the combined treatment was consistent to that obtained in the phase I study [45•], its overall toxicity appeared higher than FOLFOX treatment alone. The elevation of IFN-γ, IL-18, and granzyme B was present in the FOLFOX + pegilodecakin arm, whereas TGFβ was comparatively decreased, which accorded with immunostimulatory signals of IL-10R pathway. In addition, the upregulation of IL-18 levels correlated with better clinical outcomes in the combined treatment. It shows that the addition of pegilodecakin to FOLFOX did not significantly improve OS, PFS, or ORR in advanced gemcitabine-refractory PDAC in this phase III trial.
Conclusions and Future Directions
The IL-10 and IL-10R interaction is a key pathway in immune system that is associated with the immune response to many diseases, including tumour angiogenesis and metastasis. Its role at different stage/time during the immune response varies; as such, its regulation is a logical target for therapeutic intervention and the precise control has become one key challenge. Preclinical data clearly demonstrate a reduction in tumour burden and angiogenesis induced by pegilodecakin, which show largely increased stability than non-PEGylated IL-10 and activating tumour infiltrating CD8 + T cells. Phase 1 trials confirm the safety and tolerability of pegilodecakin as a monotherapy, or in combination with other therapeutics, including targeted therapy, chemotherapy, and anti-PD-1 inhibitors. The clinical trial results of PEG-IL-10 monotherapy or combined with other therapies have shown limited efficacy against a variety of cancer types. In addition, the use of pegilodecakin has potentiated toxicity, which made participants most likely to discontinue the trial. While PEGylation could resolve most of the toxicity problems, the binding site(s) of PEG to IL-10 is required to be characterised preclinically, as certain conformations might hinder or change the direction of receptor-ligand interaction (46), which may limit clinical outcomes. The interaction of PEG-IL-10 with IL-10 receptor 1 or/and 2 needs to be better studied, which becomes a doable task as cryo-microscopy are widely used to characterise protein–protein interaction at atomic level. Most importantly, as IL-10 can either be cancer-promoting or act as cancer therapeutics, understanding how PEG-IL-10 affects the function of other immune cells, such as monocytes/macrophages, and dendritic cells in the tumour environment will ultimately direct the way and time for the PEG-IL-10 to finally reach the clinics.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Goodnow CC, Sprent J, de St Fazekas, Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nat. 2005;435(7042):590–7.
Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361(21):2033–45.
Sturlan S, Oberhuber G, Beinhauer BG, Tichy B, Kappel S, Wang J, et al. Interleukin-10-deficient mice and inflammatory bowel disease associated cancer development. Carcinog. 2001;22(4):665–71.
• Ni G, Zhang L, Yang X, Li H, Ma B, Walton S, et al. Targeting interleukin-10 signalling for cancer immunotherapy, a promising and complicated task. Hum Vaccin Immunother. 2020;16(10):2328–32. (This article reviewed the current advances in temporal IL-10 signaling blockade enhances vaccine-induced tumor regression by CD8+ T cells and discussed the complexity of IL-10 manipulation for cancer therapy.)
Giovarelli M, Musiani P, Modesti A, Dellabona P, Casorati G, Allione A, et al. Local release of IL-10 by transfected mouse mammary adenocarcinoma cells does not suppress but enhances antitumor reaction and elicits a strong cytotoxic lymphocyte and antibody-dependent immune memory. J Immunol. 1995;155(6):3112–23.
Berman RM, Suzuki T, Tahara H, Robbins PD, Narula SK, Lotze MT. Systemic administration of cellular IL-10 induces an effective, specific, and long-lived immune response against established tumors in mice. J Immunol. 1996;157(1):231–8.
Naing A, Infante JR, Papadopoulos KP, Chan IH, Shen C, Ratti NP, et al. PEGylated IL-10 (Pegilodecakin) induces systemic immune activation, CD8+ T cell invigoration and polyclonal T cell expansion in cancer patients. Cancer Cell. 2018;34(5):775-91.e3.
Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, et al. IL-10 Directly activates and expands tumor-resident CD8+ T cells without de novo infiltration from secondary lymphoid organs. Can Res. 2012;72(14):3570–81.
Mumm John B, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. IL-10 Elicits IFNγ-dependent tumor immune surveillance. Cancer Cell. 2011;20(6):781–96.
• Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med (2020);217(1): e20190418. (This review discussed the current understanding of the regulation of IL-10 production and of the molecular pathways associated with IL-10 responses, described its nonclassic roles, and covered its therapeutic potential in the context of different diseases.)
Zheng LM, Ojcius DM, Garaud F, Roth C, Maxwell E, Li Z, et al. Interleukin-10 inhibits tumor metastasis through an NK cell-dependent mechanism. J Exp Med. 1996;184(2):579–84.
Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306.
Chen WF, Zlotnik A. IL-10: a novel cytotoxic T cell differentiation factor. J Immunol. 1991;147(2):528–34.
Lauw FN, Pajkrt D, Hack CE, Kurimoto M, van Deventer SJH, van der Poll T. Proinflammatory effects of IL-10 during human endotoxemia. J Immunol. 2000;165(5):2783–9.
Finbloom DS, Winestock KD. IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J Immunol. 1995;155(3):1079–90.
Mumm JB, Oft M. Pegylated IL-10 induces cancer immunity: the surprising role of IL-10 as a potent inducer of IFN-γ-mediated CD8(+) T cell cytotoxicity. BioEssays. 2013;35(7):623–31.
Smith SR, Terminelli C, Denhardt G, Narula S, Thorbecke GJ. Administration of interleukin-10 at the time of priming protects Corynebacterium parvum-primed mice against LPS- and TNF-alpha-induced lethality. Cell Immunol. 1996;173(2):207–14.
Huhn RD, Radwanski E, O’Connell SM, Sturgill MG, Clarke L, Cody RP, et al. Pharmacokinetics and immunomodulatory properties of intravenously administered recombinant human interleukin-10 in healthy volunteers. Blood. 1996;87(2):699–705.
Ni G, Wang T, Walton S, Zhu B, Chen S, Wu X, et al. Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol. 2015;293(2):126–9.
Rachmawati H, Beljaars L, Reker-Smit C, Van Loenen-Weemaes AM, Hagens WI, Meijer DK, et al. Pharmacokinetic and biodistribution profile of recombinant human interleukin-10 following intravenous administration in rats with extensive liver fibrosis. Pharm Res. 2004;21(11):2072–8.
Alvarez HM, So OY, Hsieh S, Shinsky-Bjorde N, Ma H, Song Y, et al. Effects of PEGylation and immune complex formation on the pharmacokinetics and biodistribution of recombinant interleukin 10 in mice. Drug Metab Dispos. 2012;40(2):360–73.
Mattos A, de Jager-Krikken A, de Haan M, Beljaars L, Poelstra K. PEGylation of interleukin-10 improves the pharmacokinetic profile and enhances the antifibrotic effectivity in CCl4-induced fibrogenesis in mice. J Control Release. 2012;162(1):84–91.
Lee S, Wylie DC, Cannon-Carlson SV. Pegylated interleukin-10. US Grant No. US7052686B2. 2000.
Oft M, Sheppard C, Mumm J, Linling W. Merck Sharp & Dohme. Pegylated IL-10 for use in treating cancer or tumor. European patent Grant No. EP2821078A1. 2015.
• Tannir NM, Papadopoulos KP, Wong DJ, Aljumaily R, Hung A, Afable M, et al. Pegilodecakin as monotherapy or in combination with anti-PD-1 or tyrosine kinase inhibitor in heavily pretreated patients with advanced renal cell carcinoma: final results of cohorts A, G, H and I of IVY Phase I study. Int J Cancer. 2021;149(2):403–8. (This study reported the phase I/Ib multi-cohort dose escalation IVY clinical trial results of pegilodecakin+ pazopanib, and final results for monotherapy and long-term follow-up with pegilodecakin + antiprogrammed cell death 1 (anti-PD-1) inhibitors.)
• Spigel D, Jotte R, Nemunaitis J, Shum M, Schneider J, Goldschmidt J, et al. Randomized phase 2 studies of checkpoint inhibitors alone or in combination with pegilodecakin in patients with metastatic NSCLC (CYPRESS 1 and CYPRESS 2). J Thorac Oncol : Off Publ Int Assoc Study Lung Cancer. 2021;16(2):327–33. (This study reported the results of randomized phase II clinical trial of checkpoint inhibitors alone or in combination with pegilodecakin in patients with metastatic non-small cell lung cancer; more definitive data on toxicity and efficacy (or lack thereof), and preliminary immune biomarker information were provided.)
• Hecht JR, Lonardi S, Bendell J, Sim HW, Macarulla T, Lopez CD, et al. Randomized phase III study of FOLFOX alone or with pegilodecakin as second-line therapy in patients with metastatic pancreatic cancer that progressed after gemcitabine (SEQUOIA). J Clin Oncol. 2021;39(10):1108–18. (The study reported that results of phase III clinical that compared efficacy and safety of adding pegilodecakin with folinic acid, fluorouracil, and oxaliplatin (FOLFOX) in patients following progression on first-line gemcitabine-containing therapy with metastatic pancreatic ductal adenocarcinoma.)
Mumm JB, Oft M. Pegylated IL-10 induces cancer immunity. BioEssays. 2013;35(7):623–31.
Blaisdell SJ, Cutler CM, Paporello BC, Ambrogelly A. Mono- and di-peg il-10 production; and uses. European Patent Grant No. EP3348281A1. 2018.
Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. IL-10 elicits IFNγ-dependent tumor immune surveillance. Cancer Cell. 2011;20(6):781–96.
Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8(1):1136.
Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22(4):433–8.
• Autio K, Oft M. Pegylated interleukin-10: clinical development of an immunoregulatory cytokine for use in cancer therapeutics. Curr Oncol Rep. 2019;21(2):19. (The reviewed discussed the development of pegilodecakin and its application from the context of preclinical investigation and the initial results of the phase I IVY studies in oncology.)
• Naing A, Wong DJ, Infante JR, Korn WM, Aljumaily R, Papadopoulos KP, et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): a multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 2019;20(11):1544–1555b. (This study assessed the safety and activity of pegilodecakin with anti-PD-1 monoclonal antibody inhibitors in patients with advanced solid tumours, including non-small-cell lung cancer, melanoma, renal cell carcinoma, triple-negative breast cancer, and bladder cancer. The Data cutoff was July 1, 2018.)
Naing A, Papadopoulos KP, Autio KA, Ott PA, Patel MR, Wong DJ, et al. Safety, antitumor activity, and immune activation of pegylated recombinant human interleukin-10 (AM0010) in patients with advanced solid tumors. J Clin Oncol : Off J Am Soc Clin Oncol. 2016;34(29):3562–9.
Berk V, Ozdemir N, Ozkan M, Aksoy S, Turan N, Inal A, et al. XELOX vs. FOLFOX4 as second line chemotherapy in advanced pancreatic cancer. Hepatogastroenterol. 2012;59(120):2635–9.
Chung JW, Jang HW, Chung MJ, Park JY, Park SW, Chung JB, et al. Folfox4 as a rescue chemotherapy for gemcitabine-refractory pancreatic cancer. Hepatogastroenterol. 2013;60(122):363–7.
Yoo C, Hwang JY, Kim JE, Kim TW, Lee JS, Park DH, et al. A randomised phase II study of modified FOLFIRI.3 vs modified FOLFOX as second-line therapy in patients with gemcitabine-refractory advanced pancreatic cancer. Br J Cancer. 2009;101(10):1658–63.
Ghosn M, Saroufim A, Kattan J, Chahine G, Nasr F, Farhat F. Sequential FOLFOX-6 and gemcitabine for locally advanced and/or metastatic pancreatic cancer. Med Oncol. 2012;29(4):2831–7.
Wainberg ZA, Feeney K, Lee MA, Muñoz A, Gracián AC, Lonardi S, et al. Meta-analysis examining overall survival in patients with pancreatic cancer treated with second-line 5-fluorouracil and oxaliplatin-based therapy after failing first-line gemcitabine-containing therapy: effect of performance status and comparison with other regimens. BMC Cancer. 2020;20(1):633.
Stein A, Binder M, Goekkurt E, Lorenzen S, Riera-Knorrenschild J, Depenbusch R, et al. Avelumab and cetuximab in combination with FOLFOX in patients with previously untreated metastatic colorectal cancer (MCRC): final results of the phase II AVETUX trial (AIO-KRK-0216). J Clin Oncol. 2020;38(4_suppl):96.
Nilsson S, Stein A, Rolfo C, Kranich AL, Mann J, Papadimitriou K, et al. Selinexor (KPT-330), an oral selective inhibitor of nuclear export (SINE) compound, in combination with FOLFOX in patients with metastatic colorectal cancer (mCRC) - final results of the phase I trial SENTINEL. Curr Cancer Drug Targets. 2020;20(10):811–7.
Sobrero A, Lonardi S, Rosati G, Di Bartolomeo M, Ronzoni M, Pella N, et al. FOLFOX or CAPOX in stage II to III colon cancer: efficacy results of the Italian three or six colon adjuvant trial. J Clin Oncol. 2018;36(15):1478–85.
• Hecht JR, Papadopoulos KP, Falchook GS, Patel MR, Infante JR, Aljumaily R, et al. Immunologic and tumor responses of pegilodecakin with 5-FU/LV and oxaliplatin (FOLFOX) in pancreatic ductal adenocarcinoma (PDAC). Invest New Drugs. 2021;39(1):182–92. (This study assessed the safety and activity of pegilodecakin + FOLFOX in patients with pancreatic ductal adenocarcinoma in an open-label phase 1b trial in the United States.)
• Hecht JR, Naing A, Falchook GS, Patel MR, Infante JR, Aljumaily R, et al. Overall survival of PEGylated pegilodecakin with 5-FU/LV and oxaliplatin (FOLFOX) in metastatic pancreatic adenocarcinoma (PDAC). 2018;36(15_suppl):4119-. (This study reported on the safety, efficacy and overall survival of AM0010 + FOLFOX as 2nd and later line treatment in pancreatic ductal adenocarcinoma.)
Fang Y, Xue J, Gao S, Lu A, Yang D, Jiang H, et al. Cleavable PEGylation: a strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Deliv. 2017;24(sup1):22–32.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cavallazzi Sebold, B., Ni, G., Li, J. et al. PEGylated IL-10: Clinical Development in Cancer Immunotherapy, Where to Go?. Curr Oncol Rep 25, 115–122 (2023). https://doi.org/10.1007/s11912-022-01355-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-022-01355-4