
Abstract
Purpose of review
Summarizing the current preclinical and clinical evidence about bystander effect of antibody–drug conjugates (ADCs) in solid tumors.
Recent findings
One of the main challenges of treating solid tumors with ADCs is the heterogeneous expression of the target antigen (Ag), which however may be overcome by the so-called bystander killing effect. This unique, but still debated, feature of certain ADCs is represented by the unintentional payload diffusion from Ag-positive tumor cells to adjacent Ag-negative tumor cells. Some pharmacological characteristics, such as a hydrophobic payload or a cleavable linker, seem to play a major role in this effect.
Summary
Abundant preclinical evidence of the bystander effect has emerged, and the clinical activity of ADCs in tumors with a heterogeneous Ag expression suggests the relevance of this feature. Additional studies are required to investigate if the bystander effect is necessary for achieving a solid activity with ADCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
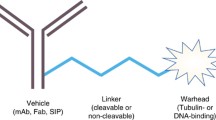
Antibody–drug conjugates (ADCs) have recently emerged as a highly effective therapeutic strategy for cancer [1••]. The embryonic idea of ADCs, namely the “magic bullet,” was firstly postulated in early 1900s by Nobel Prize Paul Ehrlich [2]. Since then, bioengineering improvements and pharmaceutical discoveries have led to the modern ADC design: a monoclonal antibody (mAb) capable of selective binding to a tumor-associated antigen and connected to a cytotoxic drug—i.e., the payload or warhead, through a chemical linker. The average number of payload molecules per ADC is named Drug-to-Antibody Ratio (DAR). Each ADC is equipped with a specific combination of these features (i.e. mAb, target molecule, linker, payload, DAR), determining the ADC-specific pharmacological and clinical properties. Consequently, complex interactions may arise between each ADC and the various constituent of the tumor microenvironment (TME) [1••, 3].
The first introduction of an ADC as treatment for solid tumors dates back to 2013, when the Food and Drug Administration (FDA) approved the human epidermal growth factor 2 (HER2)-targeted ADC ado-trastuzumab emtansine (T-DM1) for the treatment of metastatic breast cancer (BC). Since then, improvements in mAb production, linker technology, and innovative payloads determined the rise of novel ADCs, with improved clinical activity and enlarged spectrum of targetable malignancies. One of the main challenges in treating solid tumors with ADCs is the heterogeneous expression of the target antigen (Ag) in primary tumor tissues and, if present, in metastasis. Innovative cleavable linkers and hydrophobic payloads have been proposed as a possible solution, mainly because of their hypothetical major role played in the so-called “bystander killing effect.” This unique—but still debated—phenomenon is thought to improve ADC activity in cancers harboring a heterogeneous Ag expression, because of the unintentional payload diffusion from Ag-positive (Ag +) tumor cells to adjacent Ag-negative (Ag-) tumor cells [4, 5].
In this review, we summarize the current knowledge about the ADC bystander effect, discussing the challenges and the opportunities to demonstrate such activity in preclinical and clinical models.
Preclinical evidence of the bystander effect
As stated before, the bystander killing effect is the property of certain ADCs to elicit antitumor activity against cancer cells located near targeted Ag + cells, regardless of target Ag expression [4, 6]. This pharmacological characteristic seems to be strictly related to the ADC design, in particular to the biochemical features of the linker and the payload. Indeed, preclinical evidence highlights the importance of a cleavable linker that could release the payload from the Ab moiety and a hydrophobic warhead that could diffuse through the cell membrane towards neighboring cells. Mathematical models have been proposed to quantify and predict the rate and the extent of the bystander effect, mainly based on cytotoxic and in vitro studies of conjugates of the HER2-directed monoclonal antibody, trastuzumab [7]. Here we summarize the main reports of a bystander effect of different ADCs in preclinical models [4, 7].
Trastuzumab deruxtecan (T-DXd), also known as DS-8201a, is an innovative ADCs that merges the characteristics of trastuzumab with a potent topoisomerase I inhibitor, exatecan derivative (DXd), highly membrane-permeable compared to emtasine, and T-DM1 payload. The proprietary tetrapeptide-based linker of T-DXd is connected to the antibody backbone via cysteine residues, it is designed to be stable in plasma and selectively cleaved by lysosomal enzymes such as cathepsin B and L, highly expressed in tumor cells [8, 9]. On the other hand, T-DM1 is composed by trastuzumab, a non-cleavable thioether linker and DM1, an inhibitor of tubulin polymerization. Both T-DXd and T-DM1 need to recognize the HER2 extracellular domain on the surface of the target tumor cell and both need to be internalized in order to exert their antitumoral activity. However, once internalized, T-DM1 relies on the lysosomal degradation of the entire antibody–linker construct to release its payloads, resulting in the retention of charged amino acids on the payload, which affects its cell permeability.
After T-DXd is internalized into HER2 + cells and releases its payload, DXd could penetrate neighboring cells because of its high membrane permeability.
In vitro cell viability assays using a mixture of the ERBB2/HER2-amplified (HER2 +) human gastric carcinoma cells NCI-N87 and the HER2-negative (HER2-) human breast adenocarcinoma cells MDA-MB-468 demonstrated the capacity of T-DXd to kill not only the HER2 + NCI-N87 cells but also the neighbor HER2- MDA-MB-468 cells, in contrast to T-DM1 [8]. Similarly, xenograft experiments using a mixture of NCI-N87 cells and luciferase-expressing MDA-MB-468 cells validated the bystander effect of T-DXd in vivo: indeed, T-DXd reduced the luciferase signal in mice, indicating suppression of the HER2- population, while T-DM1 and another trastuzumab-based conjugated with a low-permeable payload did not. Of note, T-DXd was not effective in the HER2- cells inoculated in in a different site from the HER2 + cells, suggesting that the bystander killing effect can only be observed locally. Thus, an unspecific systemic toxicity or toxicity of normal tissues distant from the HER2 + tumor sites should be considered as low [8]. Another recent in vivo study evaluated a novel imaging system using fluorescent nanoparticles (phosphor-integrated dots, PID) as a label for immunostaining to visualize the spatial distribution of T-DXd in tumor tissues. The authors demonstrated that while trastuzumab signal overlapped with the HER2 + areas of the tumors, DXd was equally distributed in the HER2 + and HER2- regions of the tumors [10].
Trastuzumab duocarmazine, also known as SYD985, is an ADC based on trastuzumab mAb conjugated to a cleavable linker-duocarmycin payload, valine citrulline-seco DUocarmycin hydroxyBenzamide Azaindole (vc-seco-DUBA). Similarly to T-DXd, also this ADC was compared to T-DM1 in different cell lines harboring a non-homogeneous HER2 expression [11]. SYD985 and T-DM1 were marked with a fluorescent dye and their internalization was studied. The bystander killing assay was performed in co-coltures of HER2 + cell lines (SK-BR-2, SK-OV-3, MDA-MB-175-VII) with the HER2- NCI-H520 cells. NCI-H520 cells alone were found to be insensitive to SYD985 and T-DM1, but sensitive to seco-DUBA. In a co-culture containing only 20% of HER2 + cells, SYD985 was able to kill 65% of the mixed cell population, indicating a potent bystander killing effect. In the same conditions, T-DM1 was able to kill only 9% of the co-cultured cells. These findings suggest that active toxins are released after processing of SYD985 by the HER2 + cells, resulting in killing of the HER2- neighboring cells. Moreover, the antitumor activity of the ADC was tested in xenografts derived by both in cell lines and in breast cancer patient-derived xenografts, validating SYD985 activity in HER2-low and HER2-heterogeneous conditions. Pharmacokinetics assays demonstrated that HER2-mediated targeting to the tumor is essential for its antitumor activity [11]. Similar and consistent results were obtained performing in vitro and in vivo assays on epithelial ovarian cancer cells, with heterogeneous and low HER2 expression, corroborating the bystander activity of SYD985 [12].
Also other currently FDA-approved ADCs have shown bystander effect in preclinical models, for example, sacituzumab govitecan (SG) [13], tisotumab vedotin (TV) [14], enfortumab vedotin (EV) [15], and anetumab ravtansine. Anetumab ravtansine (BAY 94–9343), is a novel ADC composed by a human anti-mesothelin mAb, conjugated via a cleavable disulphide linker to microtubule inhibitor DM4. It is worth noting that although DM4 exhibit similar pharmacological and chemical characteristics to T-DM1 payload, the presence of a cleavable linker justifies its membrane permeability feature. Indeed, in vivo preclinical studies have shown the bystander property of this ADC: in particular, in patient-derived tumor models anetumab ravtansine was able to kill neighboring mesothelin-negative tumor cells, even in the presence of only 20% Ag + cells within the tumor [16].
Pharmacodynamic models using trastuzumab-based ADC combined with monometyl auristatin E (MMAE) suggested that the bystander killing effect of the ADC increases proportionally to the fraction of Ag + cells and correlates with the antigen expression levels. Another fundamental variable to consider is the time from the internalization of the ADC by the Ag + cells to diffusion of the released payload to the adjacent cells and thus to the initiation of the bystander effect. As consequence, the bystander effect can slow down over time, as the population of Ag + cells declines [7]. Finally, a predictive computational transport analysis confirmed that the bystander effect could partially (but not fully) compensate the impaired efficacy of ADCs in case of heterogeneous tumoral distribution [17].
Clinical evidence of the bystander effect
Given the amount of relevant preclinical results, it is paramount to address the question of the clinical relevance of the bystander effect. ADCs have been evaluated in many cancer subtypes in a large number of clinical trial; to date, five ADCs are approved for the treatment of solid tumors: namely T-DM1, T-DXd, EV, TV, and SG [1••, 18]. It is worth noting that the impact of a possible bystander effect, demonstrated in preclinical setting and discussed above, seems to mirror a differential clinical effect in heterogeneous Ag + tumors (Table 1).
Regarding T-DM1, its role is well established in the treatment algorithm for HER2 + (IHC 3 + or 2 + FISH test positive) metastatic BC [19, 20]. Indeed, T-DM1 has been approved in second line in HER2 + breast metastatic setting and in adjuvant treatment of HER2 + breast cancer patients not achieving a pathological complete response (pCR) after standard neoadjuvant therapy, based respectively on the results of EMILIA and KATHERINE phase III trials [21••, 22]. However, T-DM1 did not reach similar improvements in survival outcomes neither in HER2 + gastric cancer (GC) nor in HER2 + colorectal cancer (CRC), or in other histologies, as shown respectively by the results of GATSBY trial [23], HERACLES-B trial [24], or subprotocol Q of the NCI-MATCH basket trial [25]. Many hypothesis have been raised on the differential efficacy of T-DM1 across histologies, involving the different roles of HER2 as driver of oncogenesis in BC compared to other histologies, T-DM1 intracellular metabolism in GC [26], the inefficacy of the warhead, and the heterogeneous and dynamic HER2 expression profile [27, 28].
As for T-DXd, clinical data are more recent. Indeed, T-DXd is currently approved for T-DM1-pretreated BC, and received a breakthrough therapy designation by the FDA for treatment of HER2 + metastatic BC in second and later lines, due to its demonstrated activity in the phase 2 DESTINY-Breast01 clinical trial [29••] and in the DESTINY-Breast03 phase 3 clinical trial. Moreover, results of DESTINY-Gastric01 lead to the approval of T-DXd for the treatment of patients with advanced HER2 + GC who have previously received trastuzumab [30]. For CRC, the DESTINY-CRC01 phase 2 trial showed promising T-DXd activity in cohort A (HER + CRC, IHC score 2 + or 3 + and positive FISH test), while in cohort B (IHC 2 + and negative FISH result) and cohort C (IHC 1 +), no responses were observed [31].
The bystander effect could partially explain the differences in the clinical activity of the two trastuzumab-based ADCs, given the preclinical premises. This is especially true in the case of GC, where HER2 distribution is notoriously highly heterogeneous [27] and where only T-DXd was able to reach the FDA approval. Another example in this regard is given by HER2-low BC, where T-DM1 was not efficient in disease control, while novel ADCs, such as T-DXd, showed some signs of efficacy (Table 1). Whether this effect is ascribable to bystander effect is yet under investigation; nevertheless, this might be one of the major clinical arguments favoring the existence of the bystander effect. However, it should be noted that the diffusion of the payload in tissue located near the target tumor cell or into the blood stream could also hypothetically increase toxicities [3]. Indeed, the majority of novel ADCs have shown a higher rate of toxicities compared to T-DM1 [1••, 18].
Pharmacological determinants of the bystander effect
FFIGSeveral studies have tried to identify the major pharmacological determinant of the bystander killing effect of ADCs, mainly highlighting the role of a cleavable linker and a hydrophobic payload [4, 8, 16, 32,33,34]. However, some considerations should be underlined (Fig. 1).

Schematic representation of antibody–drug conjugates (ADCs) with and without bystander killing effect. In the upper side of the figure, the mechanism of action of ADCs without bystander killing effect is represented. In the lower side of the figure, the mechanism of ADCs with a potential bystander killing effect is schematized.Each prototype is characterized by the three main features thought to be mostly responsible for bystander killing. 1) Antibody-Target: the potential release of the payload before ADC internalization makes it not always essential for exerting a cytotoxic effect. However, the chemical stability of the antibody-linker bond has been studied for years in order to reach a balance between the specificity of the ADC and the risk of damage to healthy tissues, in case of premature release of the payload. 2) Linker: they are typically classified as cleavable and non-cleavable. Cleavable linkers can be subdivided into “chemically labile” (e.g., hydrazone bonds, that are pH sensitive; disulfide bonds) and “enzyme labile” (e.g., dipeptides, typically cleavable by cathepsin B; β-glucuronidase-sensitive, β-galactosidase-sensitive). Non-cleavable linkers are mainly represented by covalent bonds, such as thioether ones. They usually require intra-cellular processing (e.g., through lysosomal enzymes) for cleavage and subsequent warhead release. Consequently, for ADCs equipped with non-cleavable linkers, internalization is essential for payload release. 3) Payload: the chemical structure of the payload – its electric charge, in particular – is believed to be a critical aspect for allowing bystander killing. In fact, a payload must be able to exit the target cell by diffusing across the phospholipid bilayer, in order to kill surrounding cancer cells. To do so, the payload should be nonpolar. Conversely, in case of ADCs equipped with a polar/charged payload, such as trastuzumab emtansine, the cytotoxic drug remains trapped into the target cell. As for efficacy, ADCs exerting bystander killing effect are considered particularly indicated in case of tumors expressing the target antigen (e.g., HER2) according to a heterogeneous spectrum. The miniature figure on the left represents intra-tumor heterogeneity also in the form of sub-clonal evolution, that is typical of solid tumors. On the left side of the figure, there is the representation of the histological heterogeneity of solid tumours: green cells: tumour cells with a high expression of target antigen; blue cells: tumour with a low expression of target antigen; yellow cells: cells with no expression of target antigen. Conversely, ADCs that do not allow bystander killing should be restricted to target antigens that are expressed in a more homogeneous pattern (e.g., cluster of differentiations in hematological diseases). As for toxicity, the increased potential of bystander killing may rise concerns related to off-tumor toxicity, with damage to healthy tissues, as the main worry (miniature figure on the right). In fact, the historical early-phase clinical trials investigating the first ADCs ancestors have been interrupted due to safety concerns. Keys: Ag, antigen; DNA, deoxyribonucleic acid; ADC, antibody–drug conjugate; HER2, human epidermal growth factor receptor 2. Created with biorender.com
ADCs exists in vivo as a dynamic formulation of three components, circulating in the blood stream: the conjugate (predominant part), naked antibodies and free payload molecules. The ratio between these three components can vary, depending on incomplete conjugation during production or linker lability. After extravasation from capillaries, ADCs diffuse slowly towards the target cells and could potentially release their payload in the TME before mAb-Ag engagement, depending on redox environment, low pH and extracellular proteases. Indeed, pH-dependent linkers could naturally hydrolyze in the TME that is characterized by a lower pH compared to normal tissues due to enhanced glycolysis and lactate production. This property could explain the activity of ADCs targeting the extracellular matrix and characterized by acid-labile linkers [35, 36]. Also disulfide linkers represent a point of discussion: these are characterized by direct covalent bonds between sulfide groups that are sensitive to a reducing microenvironment, in which the release of thiols by dead tumor cells could play a major role in producing a self-amplifying effect [35, 37]. Finally, also cathepsin B cleavable linkers could be destroyed in TME, because of the hyper production of proteases by tumor cells [35].
However, after antigen binding, most ADCs are internalized via the antigen-dependent processes of endocytosis or the antigen-independent process of pinocytosis, then trafficked to endosomes, lysosomes, or caveolae [5, 32]. At this point, acid-cleavable (chemically labile, disuphide and pH-dependent) linkers are destroyed in early endosomes, while protease cleavable or enzyme (peptide based) cleavable linkers are degraded in late endosomes or lysosomes: thus, the payload is finally released and able to exert its cytotoxic activity in the intracellular space [5]. To achieve a bystander effect, the intracellular released payload must embody specific biochemical proprieties, in order to exhibit membrane permeability, diffuse through cell wall, and reach neighboring cells: it should be lipophilic, hydrophobic, and uncharged [1••, 5,6,7]. Nevertheless, ADCs exhibiting substantial extracellular payload release could potentially achieve off-target effect before internalization [35].
It should be noted that ADCs with a non-cleavable linker, such as T-DM1, have a slightly different pathway. Indeed, upon its internalization, the mAb moiety is degraded and the final payload product retains a positively charged lysine; thus, it is trapped inside target cells, and it cannot enter surrounding cells and it reaches its best activity in homogeneous Ag + expressing tumors.
Fact or fiction?
Preclinical and clinical evidence recapitulated in this manuscript solidly contribute to recognize the bystander effect as an important player in the mechanism of action of ADCs, and possibly one of the reasons leading to their expanding indications. As previously mentioned, tumors known to harbor a relevant heterogeneity in HER2 expression (e.g. GC [27], CRC [28]) did not derive a significant benefit from treatment with T-DM1, while showing solid response rates with T-DXd. Even more strikingly, novel conjugates, such as T-DXd, have shown response rates in the range of 30–40% in breast tumors with low HER2 expression, a setting in which T-DM1 did not show to be active. At the same time, examples exist of novel ADCs with an increased activity compared to T-DM1, despite the absence of the bystander effect. This is the case, for instance, of ARX788, composed of a modified trastuzumab with a p-acetylphenylalanine residue linked to the microtubule inhibitor amberstatin through a site-specific conjugation, which leads to a highly stable compound and a tightly controlled DAR. Such stability is thought to reduce the shedding of payload in the circulation, thus improving the safety profile and the delivery of cytotoxic to cancer cells. At the same time, the very design of this conjugate prevents the induction of the bystander effect, restricting the delivery of payload to the targeted cells [38]. Nonetheless, the compound showed a preliminary but impressive response rate of 74% in patients with T-DM1-pretreated HER2-positive advanced BC [39], and preclinical evidence suggest it may be also harbor activity against HER2-low BC [38].
The activity of ARX788, despite the absence of a bystander effect, suggests caution in attributing to the bystander effect the sole reason for the current development of novel ADCs. Indeed, these are complex pharmaceuticals, whose activity is determined by a multiplicity of factors beyond the linking technology or the membrane permeability of the payload [1••]. A rapidly enlarging landscape of clinical trial testing novel ADCs is expected to clarify the importance of the bystander effect, informing the design of the next generation of ADCs.
Conclusion
ADCs represent a highly active class of drugs for the treatment of cancer. Abundant preclinical evidence on the importance of the bystander effect has emerged, and the clinical activity of novel conjugates in tumors with a heterogeneous expression of the targeted Ag suggests the relevance of this feature. However, more stable ADCs, not able to elicit the bystander effect, are also being pursued for the treatment of cancer, recently demonstrating promising clinical efficacy and preclinical activity even in HER2-low tumors. Limited data are available on the possible and plausible role of the bystander effect in off-target toxicities. Additional studies are required to understand if the bystander effect is a necessary feature for achieving widely active ADCs or if it is only one of the several factors to be considered in the design of personalized ADCs.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
•• Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat Rev Clin Oncol [Internet]. 2021 Jun 8;18(6):327–44. Available from: http://www.nature.com/articles/s41571-021-00470-8. This is a comprhensive review on the topic of ADCs.
Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer [Internet]. 2008;8(6):473–80. Available from: http://www.nature.com/articles/nrc2394.
Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs [Internet]. 2016 May 18;8(4):659–71. Available from: http://www.tandfonline.com/doi/full/10.1080/19420862.2016.1156829.
Kovtun Y V., Audette CA, Ye Y, Xie H, Ruberti MF, Phinney SJ, et al. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res [Internet]. 2006 Mar 15;66(6):3214–21. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-05-3973.
Kovtun Y V., Goldmacher VS. Cell killing by antibody–drug conjugates. Cancer Lett [Internet]. 2007 Oct;255(2):232–40. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0304383507001991.
Li F, Emmerton KK, Jonas M, Zhang X, Miyamoto JB, Setter JR, et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res [Internet]. 2016;76(9):2710–9. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-15-1795.
Singh AP, Sharma S, Shah DK. Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J Pharmacokinet Pharmacodyn [Internet]. 2016;;43(6):567–82. Available from: http://springerlink.bibliotecabuap.elogim.com/10.1007/s10928-016-9495-8.
Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of <scp>DS</scp> ‐8201a, a novel anti‐human epidermal growth factor receptor 2 antibody–drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci [Internet]. 2016;107(7):1039–46. Available from: https://onlinelibrary.wiley.com/doi/10.1111/cas.12966.
Aggarwal N, Sloane BF. Cathepsin B: Multiple roles in cancer. PROTEOMICS - Clin Appl [Internet]. 2014;8(5–6):427–37. Available from: https://onlinelibrary.wiley.com/doi/10.1002/prca.201300105.
Suzuki M, Yagishita S, Sugihara K, Ogitani Y, Nishikawa T, Ohuchi M, et al. Visualization of Intratumor Pharmacokinetics of [fam-] Trastuzumab Deruxtecan (DS-8201a) in HER2 Heterogeneous Model Using Phosphor-integrated Dots Imaging Analysis. Clin Cancer Res [Internet]. 2021;27(14):3970–9. Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-21-0397.
van der Lee MMC, Groothuis PG, Ubink R, van der Vleuten MAJ, van Achterberg TA, Loosveld EM, et al. The preclinical profile of the duocarmycin-based HER2-targeting ADC SYD985 predicts for clinical benefit in low her2-expressing breast cancers. Mol Cancer Ther [Internet]. 2015;14(3):692–703. Available from: http://mct.aacrjournals.org/lookup/doi/10.1158/1535-7163.MCT-14-0881-T.
Menderes G, Bonazzoli E, Bellone S, Black J, Altwerger G, Masserdotti A, et al. SYD985, a novel duocarmycin-based HER2-targeting antibody-drug conjugate, shows promising antitumor activity in epithelial ovarian carcinoma with HER2/Neu expression. Gynecol Oncol [Internet]. 2017;146(1):179–86. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0090825817308041.
Perrone E, Lopez S, Zeybek B, Bellone S, Bonazzoli E, Pelligra S, et al. Preclinical activity of sacituzumab govitecan, an antibody-drug conjugate targeting trophoblast cell-surface antigen 2 (Trop-2) linked to the active metabolite of irinotecan (SN-38), in ovarian cancer. Front Oncol [Internet]. 2020;10. Available from: https://www.frontiersin.org/article/10.3389/fonc.2020.00118/full.
Alley SC, Harris JR, Cao A, Heuvel EG den, Velayudhan J, Satijn D, et al. Abstract 221: Tisotumab vedotin induces anti-tumor activity through MMAE-mediated, Fc-mediated, and Fab-mediated effector functions in vitro. In: Experimental and Molecular Therapeutics [Internet]. American Association for Cancer Research; 2019. p. 221–221. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/1538-7445.AM2019-221.
Liu BA, Olson D, Snead K, Gosink J, Tenn E-M, Zaval M, et al. Abstract 5581: Enfortumab vedotin, an anti-Nectin-4 ADC demonstrates bystander cell killing and immunogenic cell death anti-tumor activity mechanisms of action in urothelial cancers. In: Immunology [Internet]. American Association for Cancer Research; 2020. p. 5581–5581. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/1538-7445.AM2020-5581.
Golfier S, Kopitz C, Kahnert A, Heisler I, Schatz CA, Stelte-Ludwig B, et al. Anetumab ravtansine: a novel mesothelin-targeting antibody–drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther [Internet]. 2014;13(6):1537–48. Available from: http://mct.aacrjournals.org/lookup/doi/10.1158/1535-7163.MCT-13-0926.
Khera E, Cilliers C, Bhatnagar S, Thurber GM. Computational transport analysis of antibody-drug conjugate bystander effects and payload tumoral distribution: implications for therapy. Mol Syst Des Eng [Internet]. 2018;3(1):73–88. Available from: http://xlink.rsc.org/?DOI=C7ME00093F.
Tarantino P, Carmagnani Pestana R, Corti C, Modi S, Bardia A, Tolaney SM, et al. Antibody–drug conjugates: Smart chemotherapy delivery across tumor histologies. CA Cancer J Clin [Internet]. 2021; Available from: https://onlinelibrary.wiley.com/doi/10.3322/caac.21705
Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet [Internet]. 2021;397(10286):1750–69. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0140673620323813.
Gennari A, André F, Barrios CH, Cortés J, de Azambuja E, DeMichele A, et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann Oncol [Internet]. 2021;32(12):1475–95. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0923753421044987.
•• Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N Engl J Med [Internet]. 2012;367(19):1783–91. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1209124. This is the EMILIA trial, that led to the approval of the first ADC for the treatment of solid tumours.
von Minckwitz G, Huang C-S, Mano MS, Loibl S, Mamounas EP, Untch M, et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N Engl J Med [Internet]. 2019;380(7):617–28. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1814017.
Thuss-Patience PC, Shah MA, Ohtsu A, Van Cutsem E, Ajani JA, Castro H, et al. Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): an international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol [Internet]. 2017;18(5):640–53. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1470204517301110.
Sartore-Bianchi A, Lonardi S, Martino C, Fenocchio E, Tosi F, Ghezzi S, et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: the phase II HERACLES-B trial. ESMO Open [Internet]. 2020;5(5):e000911. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2059702920327150.
Jhaveri KL, Wang XV, Makker V, Luoh S-W, Mitchell EP, Zwiebel JA, et al. Ado-trastuzumab emtansine (T-DM1) in patients with HER2-amplified tumors excluding breast and gastric/gastroesophageal junction (GEJ) adenocarcinomas: results from the NCI-MATCH trial (EAY131) subprotocol Q. Ann Oncol [Internet]. 2019;30(11):1821–30. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0923753420325904.
Wang H, Wang W, Xu Y, Yang Y, Chen X, Quan H, et al. Aberrant intracellular metabolism of T-DM1 confers T-DM1 resistance in human epidermal growth factor receptor 2-positive gastric cancer cells. Cancer Sci [Internet]. 2017;108(7):1458–68. Available from: https://onlinelibrary.wiley.com/doi/10.1111/cas.13253.
Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet [Internet]. 2020;396(10251):635–48. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0140673620312885.
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet [Internet]. 2019;394(10207):1467–80. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0140673619323190.
•• Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med [Internet]. 2020;382(7):610–21. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1914510. These are the results of DESTINY-Breast01.
Shitara K, Bang Y-J, Iwasa S, Sugimoto N, Ryu M-H, Sakai D, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N Engl J Med [Internet]. 2020;382(25):2419–30. Available from: http://www.nejm.org/doi/10.1056/NEJMoa2004413.
Siena S, Di Bartolomeo M, Raghav K, Masuishi T, Loupakis F, Kawakami H, et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): a multicentre, open-label, phase 2 trial. Lancet Oncol [Internet]. 2021;22(6):779–89. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1470204521000863.
Erickson HK, Park PU, Widdison WC, Kovtun Y V., Garrett LM, Hoffman K, et al. Antibody-Maytansinoid Conjugates Are Activated in Targeted Cancer Cells by Lysosomal Degradation and Linker-Dependent Intracellular Processing. Cancer Res [Internet]. 2006;66(8):4426–33. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-05-4489.
Masuda S, Miyagawa S, Sougawa N, Sawa Y. CD30-targeting immunoconjugates and bystander effects. Nat Rev Clin Oncol [Internet]. 2015;12(4):245–245. Available from: http://www.nature.com/articles/nrclinonc.2014.159-c1.
Li JY, Perry SR, Muniz-Medina V, Wang X, Wetzel LK, Rebelatto MC, et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell [Internet]. 2016;29(1):117–29. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1535610815004729.
Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer [Internet]. 2017;117(12):1736–42. Available from: http://www.nature.com/articles/bjc2017367.
Sharkey RM, Karacay H, Govindan S V., Goldenberg DM. Combination Radioimmunotherapy and Chemoimmunotherapy Involving Different or the Same Targets Improves Therapy of Human Pancreatic Carcinoma Xenograft Models. Mol Cancer Ther [Internet]. 2011;10(6):1072–81. Available fromhttp://mct.aacrjournals.org/lookup/doi/10.1158/1535-7163.MCT-11-0115.
Bernardes GJL, Casi G, Trüssel S, Hartmann I, Schwager K, Scheuermann J, et al. A Traceless Vascular-Targeting Antibody-Drug Conjugate for Cancer Therapy. Angew Chemie Int Ed [Internet]. 2012;51(4):941–4. Available from: https://onlinelibrary.wiley.com/doi/10.1002/anie.201106527.
Skidmore L, Sakamuri S, Knudsen NA, Hewet AG, Milutinovic S, Barkho W, et al. ARX788, a Site-specific Anti-HER2 Antibody–Drug Conjugate, Demonstrates Potent and Selective Activity in HER2-low and T-DM1–resistant Breast and Gastric Cancers. Mol Cancer Ther [Internet]. 2020;19(9):1833–43. Available from: http://mct.aacrjournals.org/lookup/doi/10.1158/1535-7163.MCT-19-1004.
Hurvitz SA, Park H, Frentzas S, Shannon CM, Cuff K, Eek RW, et al. Safety and unique pharmacokinetic profile of ARX788, a site-specific ADC, in heavily pretreated patients with HER2-overexpresing solid tumors: Results from two phase 1 clinical trials. J Clin Oncol [Internet]. 2021;39(15_suppl):1038–1038. Available from: https://ascopubs.org/doi/10.1200/JCO.2021.39.15_suppl.1038.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
PT served as advisor/consultant for AstraZeneca. GC received honoraria for speaker, consultancy, or advisory rule from AstraZeneca, Roche, Pfizer, Novartis, Seattle Genetics, Lilly, Ellipses Pharma, Foundation Medicine, Daiichi Sankyo, and Samsung. The other authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Breast Cancer
Rights and permissions
About this article

Cite this article
Giugliano, F., Corti, C., Tarantino, P. et al. Bystander effect of antibody–drug conjugates: fact or fiction?. Curr Oncol Rep 24, 809–817 (2022). https://doi.org/10.1007/s11912-022-01266-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-022-01266-4