
Abstract
Purpose of Review
Since its initial approval in 1997, rituximab has revolutionized the treatment of CD20-positive lymphoproliferative disorders. Now, over two decades later, second-generation molecules are emerging that may have key biological advantages compared to rituximab, as well as biosimilars that may be more cost-effective. Clinicians, health policy makers, and payers will now need to critically appraise the available evidence for these competitors and decide which anti-CD20 to use.
Recent Findings
Evidence has emerged directly comparing rituximab IV to a subcutaneous preparation, and head-to-head comparisons of rituximab versus next-generation anti-CD20 monoclonal antibodies have also been published. Trials comparing rituximab with newly developed biosimilars have also allowed for registration of these agents.
Summary
In this review, we will present an overview of anti-CD20 monoclonal antibody development, discuss the mechanistic and clinical evidence for rituximab, as well as the novel compounds, and provide commentary on the possible advantages and limitations of these agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction—The History of Anti-CD20 Monoclonal Antibody Development
The identification of the CD20 antigen in 1979 (then called B1) was the first step in what would become a therapeutic milestone [1, 2]. This glycosylated phosphoprotein is expressed on the surface of developing B cells but not the early progenitors nor mature plasma cells, and it was hypothesized (and later confirmed) that depleting these intermediate stage B cells in humans would be well tolerated without significant side effects [3]. Despite decades of study, the exact function of CD20 remains somewhat poorly understood. It is thought to play a role in calcium transport and is a member of the membrane spanning 4-A family with two small extracellular loops [4]. It is expressed at various levels by almost all B cell malignancies and is largely limited to B cells, identified early as a potential target for monoclonal antibody (mAb) therapy. Although the concept of immunotherapy had been around for almost a century, it took the Nobel-prize winning work of Köhler and Milstein in 1975 to begin this therapeutic revolution. They generated the first hybridoma cell lines capable of producing mAbs by immunizing mice against sheep cells followed by isolation of B-lymphocytes from the murine spleens and subsequent fusion of those cells with a myeloma cell line [5]. This was followed in 1980 by a proof of principle serotherapeutic trial in which a patient with multiply relapsed poorly differentiated “lymphocytic lymphoma” was treated with a murine anti-CD20 monoclonal antibody. A transient response was observed—with reduction in circulating cells—and the quest to mass-produce anti-CD20 mAbs began [6].
The first mAbs produced were murine. Early experience was disappointing with responses transient, half-lives short, and the formation of human anti-mouse antibodies’ limited efficacy. Monoclonal antibodies are very large and complex molecules and variably glycosylated (which has impact on their function), and the originally produced hybridomas could not generate sufficient quantities for cost-effective therapy. Two main technical advances were required to overcome these limitations: first, recombinant DNA technology to make the mouse antibodies more like human ones and second, engineered mammalian cells able to mass-produce sufficient quantities of the desired antibody to make treatment commercially viable (Fig. 1) [7].

Manufacture of monoclonal antibodies. 1. Immunize mouse (or other animal) with antigen of interest (human anti-CD20). 2. Isolate murine plasma cells expressing antibody targeting antigen. 3. Fuse with myeloma immortalized line of cells. 4. Generate hybridomas secreting the mAb. 5. Isolate clone producing the optimal antibody. 6. Assay antibody to ensure reactivity. 7. Clone antibody gene. 8. Express in Chinese hamster ovary cells. 9. Cell culture. 10. Bioreactor. 11. Harvest, filtration, purification, and quality control
The first success was in making “chimeric” antibodies—from the Greek mythological monster made up of many parts (“chimera”)—these antibodies had murine variable regions that provided the antigen specificity and human constant regions that were able to interact with human host effector cells and complement. The human Fc portion also prolonged the half-life by interacting with receptors on endothelial cells lining the human vasculature [8, 9]. Subsequent advancements have led to the ability to produce “humanized” (less mouse-like) and fully human antibodies (Fig. 1).
The recombinant DNA that codes for the protein must be integrated into mammalian cells that can secrete large amounts of the desired antibody. It is important to bear in mind that the choice of mammalian cell used has impact on the composition of the N-linked sugars on the molecule which can impact the pharmacological properties of the mAb generated. A group at IDEC pharmaceuticals manipulated Chinese hamster ovary cells by genomic amplification with a linked dihydrofolate reductase gene and selection with competitive inhibition by methotrexate [7]. This technology was up-scalable. The Fc region of the chimeric antibody they produced was glycosylated in a way that allowed for interaction with human effector functions. This antibody, IDEC-C2B8, was later renamed rituximab.
Rituximab
Since its initial approval in 1997, rituximab has revolutionized the treatment of B cell malignancies [10••]. It is used as a monotherapy and in combination, at induction and relapse, and also as maintenance [10••]. Over the course of two decades, clinical trials were able to demonstrate clinically meaningful differences in progression-free and, in some cases, overall survival. Its favorable side effect profile has been well established [11], allowing for safe and tolerable combination therapy with both chemotherapeutics and novel agents. The success of rituximab spurred the development of second-generation molecules, aimed to improve efficacy, as well as biosimilars that may be more cost-effective. With these emerging compounds, it is time to ask what is the role of these newer agents?
Subcutaneous Rituximab
Rituximab was initially formulated as an intravenous (IV) infusion, due to the large volume required to administer doses considered therapeutic. By concentrating the solution more than 12-fold and co-administering with recombinant human hyaluronidase (rHuPH20), an enzyme that reduces resistance in the tissue by transiently depolymerizing interstitial hyaluronan, a subcutaneous (SC) preparation of the same active molecule was made possible [12•, 13]. As the IV infusion is typically administered over a period of 1.5–6 h, the SC preparation is an attractive alternative, with shortened administration time (5–7 min) and fixed dosing (1400 mg for NHL or 1600 mg for CLL) reducing mixing time as well as waste [12•, 14].
As the active molecule is the same, the studies conducted for regulatory approval were designed to demonstrate pharmaco-equivalence. The objective of the initial dose-finding phase Ib SparkThera study was to determine a SC dose that would yield a similar trough concentration as the IV at the standard dose of 375 mg/m2 and a starting point for the CLL dose-finding study, SAWYER [15, 16]. The data generated suggested that even across all body surface area subgroups, the selected doses of 1400 mg for NHL and 1600 mg for CLL would result in adequate exposure compared with the IV dosing.
Pharmaco-equivalence and similar response rates compared to the IV version have been demonstrated across a number of settings: FL (SABRINA), DLBCL (MabEASE), and CLL (SAWYER), and this preparation has approval for use as induction, at relapse (FL and CLL) or as maintenance (FL) [17,18,19]. As the initial dose was still given IV across all the SC studies, the incidence of first-dose infusion-related reactions is unchanged, and the only significant difference in safety profile was an increase in administration-site and local cutaneous reactions [12•, 20].
While not intended to be better, in a prospective, randomized, open-label, crossover study involving 743 patients (PrefMab), 77–84% of patients reported a preference for the SC over the IV preparation. Identified reasons included “less time in clinic” and “feels more comfortable” [21]. There have also been numerous studies evaluating the impact on healthcare resource utilization. The SC preparation has been shown to reduce chair-time, preparation time, prescribing time, and waste [22,23,24,25]. A small pilot study even demonstrated the feasibility of self-administration as an outpatient with further reduction in costs [26]. Prescribers may also be reassured by the fact that SC rituximab is the same active molecule that underwent decades of evaluation in multiple clinical trials.
Next-Generation Development—Overcoming Rituximab Resistance
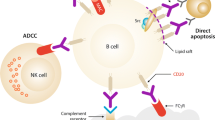
Investigation into the mechanism of action of rituximab led to the observation that anti-CD20 mAbs can largely be separated into two groups [27,28,29]. “Type I” mAbs comprise the majority (including rituximab), cause the CD20 molecule to cluster into lipid rafts on the membrane, and activate complement-dependent cytotoxicity (CDC) and possibly some degree of caspase-dependent direct cell death (DCD) (Fig. 2). The Fc portion of these mAbs is also capable of recruiting an effector immune response—antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent phagocytosis (ADP). By contrast, “Type II” mAbs do not cause clustering of CD20 into rafts, they are relatively ineffective at activating complement but evoke more potent caspase-independent DCD and are better at recruiting ADCC and ADP (Table 1) [30, 31, 34,35,36,37, 42, 43].

Type I mAbs induce the formation of lipid rafts of CD20 and bind between tetramers, cause more complement-dependent cytotoxicity (CDC) and less direct cell death (DCD). By contrast, type II mAbs do not cause the formation of lipid rafts, nor do they significantly activate complement. They do generate more non-caspase-dependent DCD. The effect of low affinity polymorphisms of the Fcgamma receptor expressed on effector cells is thought to reduce antibody-dependent cellular cytotoxicity (ADCC). The impact of glyco-engineering the Fc portion of mAbs is to more effectively engage ADCC and antibody-dependent phagocytosis (ADP) facilitating binding and effector cell recruitment even in those patients with low affinity Fcgamma polymorphisms
Despite extensive use of rituximab in the treatment of B cell–derived neoplasms, there are patients that fail to respond to initial therapy or relapse sooner than might be expected. Rituximab resistance has been somewhat arbitrarily defined lack of response during or relapse within 6 months of a rituximab-containing regimen [44]. Drivers of resistance have been extensively studied and may be tumor or host related. Exhaustion of complement, trogocytosis, lower affinity Fcgamma receptor polymorphisms, downregulation of CD20, upregulation of anti-apoptotic proteins, and host effector cell exhaustion have all been incriminated [45,46,47,48]. Strategies to overcome these mechanisms of resistance led to the development of next-generation molecules, aiming to be better than rituximab.
Ofatumumab
A fully human mAb directed against a unique epitope of the CD20 molecule, ofatumumab (OFA) was generated with a transgenic mouse and hybridoma technology [32, 49]. Postulated advantages over rituximab included the fact that it was fully human (thus less immunogenic), had a very low off-rate, and by virtue of its binding site being in closer proximity to the membrane could activate CDC more readily—with less reliance on effector mechanisms [32, 50]. In vitro, OFA demonstrated greater activity against CLL cells than rituximab and could lyse rituximab-resistant cells that expressed low levels of CD20.
It was initially approved in patients with relapsed and refractory (RR) CLL that had relapsed during or within 6 months of treatment with either a fludarabine-containing regimen or alemtuzumab. The investigator-determined overall response rate (ORR) in those refractory to fludarabine and alemtuzumab was 42%, with a median duration of response of 6.5 months [49, 51]. However, OFA in this population was later shown to be inferior to ibrutinib (IBR) with long-term follow-up of the RESONATE study (OFA vs IBR) demonstrating significantly prolonged progression-free survival (PFS) (3-year PFS, 59% vs 3%), and despite a 68% crossover, improved OS for IBR [52, 53].
In treatment-naïve patients unfit for fludarabine, OFA combined with chlorambucil (OFA+CLB) has proven superior to chlorambucil (CLB) alone [54]. It also prolonged PFS in R/R CLL compared with observation alone in a maintenance setting [55]. However, a recent meta-analysis suggested it may be a less cost-effective choice in the current era [56, 57].
So is ofatumumab superior to rituximab? Some debate exists over the relative importance of CDC in humans, with in vivo studies in transgenic mice suggesting effector-mediated mechanisms are the most important [58, 59••]. Available evidence from direct head-to-head comparisons with rituximab suggest that the advantages of OFA do not appear to translate into clinical superiority. In the ORCHARRD trial, patients with relapsed DLBCL were treated with DHAP (dexamethasone, high-dose cytarabine, cisplatin) combined with either OFA or RTX, and no advantage was seen between groups [60]. Similarly, no difference was observed when patients with relapsed iNHL were randomized to single-agent OFA or RTX [61]. Both trials were closed early for futility.
Obinutuzumab
The only type II anti-CD20 mAb currently marketed, obinutuzumab (OBZ) was engineered to overcome proposed mechanisms of rituximab resistance. A humanized, rather than fully human, molecule, its type II properties together with a modified elbow-hinge region cause greater non-caspase-dependent DCD [62,63,64]. In contrast to RTX and OFA, it does not cause CD20 to form lipid rafts and does not significantly activate complement [27, 30, 31, 37]. Manufactured in cells with overexpression of glycosylation enzymes, the resultant antibody also has non-fucosylated sugars on the Fc portion, which improves binding to the Fc receptors and can evoke more potent responses from the host immune system (Fig. 2) [62, 65]. When compared to RTX in vitro, OBZ demonstrated greater DCD, ADCC, and ADP—regardless of the Fcgamma receptor phenotype [37, 42, 43, 62].
After demonstrating activity in single-arm studies of patients with R/R CD20–positive malignancies, OBZ underwent direct comparison with RTX across a variety of settings [66,67,68]. As monotherapy in R/R iNHL, investigator-assessed ORR favored OBZ over RTX (44.6% vs 33.3%; p = 0.08); CR rates were higher for OBZ (41.9% vs 22.7%, p = 0.006), but no difference in PFS was observed between groups [69]. In patients with CLL and comorbidities, compared to RTX+CLB, OBZ+CLB demonstrated significant prolongation of PFS (28.9 vs 15.7 months; p < 0.0001) and OS (median not reached vs 73.1 months; HR, 0.76; p = 0.02) [70, 71].
In patients with treatment naïve, advanced stage symptomatic FL, OBZ-chemo vs RTX-chemo (followed by maintenance with the randomized antibody in responding patients) prolonged investigator-assessed 3-year PFS (82% vs 75%; p = 0.002) although end-of-induction response and OS were not statistically different between groups [72, 73]. The benefit of OBZ over RTX in prolonging PFS and time-to-next-anti-lymphoma treatment (TTNALT) was seen regardless of chemotherapy backbone used (bendamustine, CHOP, or CVP); however, a higher proportion of fatal adverse events was reported in patients treated with bendamustine. The majority of these events occurred during the maintenance phase, in patients more likely to be older or with comorbidities, and bendamustine induction was associated with greater reduction in CD4+T-cells [72]. These results have led some to question the use of maintenance following bendamustine induction with any anti-CD20+ mAb and also the use of bendamustine in older, frailer patients [74]. For patients with previously untreated DLBCL, two phase III randomized trials (GOYA and GAINED) failed to demonstrate any advantage of OBZ over RTX combined with standard chemotherapy [75, 76].
The rationale for the selected dosing schedule of obinutuzumab has been extensively reviewed elsewhere [46, 77]. In preclinical experiments, obinutuzumab was superior to rituximab in causing DCD, ADCC, and lymphocyte depletion when identical concentrations of both agents were used. Further increasing the concentration of rituximab demonstrated a plateau in activity, which was not observed for obinutuzumab [37, 62, 78]. Furthermore, at saturating concentrations, obinutuzumab bound to B cells at levels approximately 50% less than rituximab. Taken together, preclinical evidence might suggest that for an equal antigenic mass, less obinutuzumab is necessary to evoke cytotoxicity and less is taken up by the antigenic “sink” of the tumor compared with rituximab [46, 62]. Regardless, the dosing strategy for obinutuzumab does result in the administration of more antibodies (Table 2) which is of similar molecular weight to rituximab [46]. If dose does matter, this would confer an advantage to obinutuzumab in the head-to-head comparative trials. However, as discussed, it may be the case that rituximab, more than obinutuzumab, has a dose-related ceiling of efficacy.
Is obinutuzumab better than rituximab? It does appear to be a more potent antibody with higher rates of minimal residual disease (MRD) negativity observed compared to RTX in both patients with CLL (CLL-11) and FL (GALLIUM) [70, 81]. Capitalizing on the ability to generate deep responses, combinations of OBZ with venetoclax (Bcl2 inhibitor) in CLL patients have demonstrated very impressive results with ORR ranging from 90 to 100% and MRD-negativity rates in peripheral blood of 87–92% [82, 83]. Further analysis of GALLIUM in FL suggests that OBZ-chemo reduces the risk of early progression compared with RTX-chemo by 34% at 2 years, potentially improving outcomes for the highest risk “POD-24” cohort that has been well described [84, 85]. There also may be a subset of DLBCL patients with increased expression of germinal-center genes that benefit from OBZ more than RTX [86]. However, the increase in adverse events (in particular infusion-related reactions and cytopenias) observed with OBZ compared with RTX along with the inevitable increase in cost (compared to subcutaneous rituximab or biosimilars) has led to great debate regarding its role.
Ublituximab
Like rituximab and ofatumumab, ublituximab (UBX) has type I properties, but targets a different epitope of the CD20 molecule and has, similar to obinutuzumab, been glyco-engineered [87]. Pre-clinically, UBX demonstrates similar levels of CDC and DCD compared with RTX, but enhanced ADCC was demonstrated—even in RTX-resistant cell lines [87,88,89]. Initial dose-finding investigation was promising, and the 900 mg dose was selected to move forward in a number of studies investigating its use combined with novel agents in R/R CD20+ lymphoproliferative neoplasms [90, 91].
Combination with a next-generation oral PI3Kδ/CK1ε inhibitor, umbralisib (TGR-1202) as well as ibrutinib (a chemotherapy-free “triplet”) has been evaluated in a phase 1 setting enrolling patients with R/R NHL [92]. The combination of umbralisib (TGR-1202) and UBX (“U2”) is also under evaluation in the three-arm UNITY-NHL phase 2b randomized trial (NCT02793583). The global registration directed UNITY-CLL phase 3 randomized trial (NCT02612311) is enrolling both treatment-naïve and R/R CLL patients and randomizing to U2 or OBZ plus CLB, similar to the CLL-11 trial. This agent has not yet received regulatory approval.
Is ublituximab better than rituximab? In vitro evidence suggests it may harbor some advantage, but without any direct comparisons to rituximab in the clinical setting, it may prove challenging to answer this question with any certainty.
Biosimilars—Making a Cost-Effective Alternative
With an estimated global expenditure of US$100 billion per annum on anti-cancer medicines, predicted to rise to $150 billion by 2020, comes a universal push to reduce spending by facilitating biosimilars [93]. The Food and Drug Administration (FDA) recently released an action plan to increase the availability of biosimilars, given that biologics represent 70% of the increase in drug spending between 2010 and 2015 and the global biosimilars market is predicted to reach US$35 billion by 2020 [94, 95].
In order to claim that a molecule is biosimilar to the authorized reference product, it must be shown to have very similar structure, biological activity, efficacy, safety, and immunogenicity profile [96]. The European Medicines Association (EMA), which has been approving biosimilars since 2006, recommends rigorous evaluation—both pre- and post-approval [97]. Pre-clinically, high similarity in chemical and biological characteristics must be demonstrated. Clinical similarity then must be evaluated against the comparator directly. Trials should be designed to prove non-inferiority of both efficacy and safety against the reference product. There is also a post-approval commitment required to generate ongoing “real-world” evidence. Once biosimilarity has been demonstrated, data may be extrapolated to other indications already approved for the reference product, theoretically avoiding unnecessary repetition of trials already performed.
It is important to highlight several challenges regarding the development of biosimilars. As outlined earlier, the manufacturing process for these large molecules is highly complex, and differences in structural features such as fucosylation and protein folding can have significant impact on biological activity [96, 98••]. Even for rituximab, small changes in the glycosylation pattern have appeared over time and changes in the manufacturing process (site transfers, scale changes, etc.) have been found to alter the commercially available product [99].
Proprietors of the patented reference molecule are not obliged to disclose the exact details of their production process. Therefore, biosimilar manufacturers must develop their own methods, then demonstrate that the generated biosimilar exhibits comparable structural and functional properties prior to commencing further clinical evaluation [98••]. This does allow the biosimilar companies to capitalize on technical advances since the reference product was first developed.
The confirmatory clinical trials that support the approval of a biosimilar have been allowed to use ORR as a surrogate endpoint [97]. In the past, trials involving RTX or next-generation mAbs have used a variety of clinical endpoints including PFS, event-free survival, TTNALT, and OS. ORR is understandably an attractive endpoint—outcomes may be rapidly and easily obtained, expediting access to market for the biosimilar. Careful post-marketing follow-up will be crucial to ensure that response rates for biosimilars translate into meaningful longer term clinical outcomes. Finally, not all countries have the same regulatory approval process which means that not all biosimilars will be subjected to the same degree of pre- and post-approval scrutiny.
GP2013 (Rixathon)
GP2013 is a rituximab biosimilar that has been developed in accordance with EMAs guidance, including non-clinical and preclinical investigations and subsequent trials in rheumatoid arthritis and FL. In treatment-naïve–advanced-stage FL patients, equivalence was demonstrated between rixathon-CVP and RTX-CVP with ORRs 87% vs 88% respectively [100]. Follow-up was quite short at only 11.6 months. Safety, tolerability, and immunogenicity profiles were similar between groups. This was sufficient for EMA to issue broad approval for the biosimilar across all indications currently held for rituximab—although with the caveat that it “is subject to additional monitoring” [101].
CT-P10 (Truxima)
Similar to GP2013, CT-P10 has demonstrated its similarity with RTX pre-clinically and also underwent phase III randomized evaluation in previously untreated FL patients with advanced-stage disease. In this smaller study of 140 patients, ORR in the efficacy population was non-inferior at 97% in CT-P10 plus CVP arm vs 93% in the RTX-CVP arm. Again, no differences in pharmacokinetic properties, safety, or immunogenicity were demonstrated, and this has also led to a broad approval in Europe similar to rixathon [102].
Reditux
This intended biosimilar has been marketed since 2007 in India and was produced by the Indian generic manufacturer, Dr. Reddy’s. Its launch price was 50% less than the originator and within 3 years of approval, had increased access for patients in India by sixfold [103]. India is a semi-regulated pharmaceutical market, and clinical trials required for approval of biosimilars only require evidence of safety and biologic equivalence. Reditux does have a different chromatography profile than rituximab and production differences between the two molecules exist. Approval in India was based on a single-arm clinical trial involving 17 patients [103, 104]. More recently, a limited retrospective evaluation has suggested a similar pharmacokinetic profile, toxicity, and response rates in DLBCL patients [105,106,107]. Since the launch of reditux in India, the manufacturer sought approval in other semi-regulated countries like Peru [103]. At the current time, reditux cannot seek approval in Europe or North America because of respective regulations governing biosimilars.
What is the role of biosimilars? If truly bioequivalent, the main advantage of biosimilars is cost. In an analysis performed to investigate the impact of replacing RTX with CT-P10 (truxima), assuming a reduction in acquisition cost by just 30%, data suggested savings across Europe could exceed €90 million within 12 months and projected a €570 million saving by 3 years, potentially giving an additional 47,695 patients’ access to treatment [108]. 43.3% of the savings would come from the treatment of NHL and 19.8% from treating CLL. Authors noted their estimate for the price of CT-P10 might be conservative, with greater savings possible based on increasing competition and associated reductions in price.
Conclusion
Since its approval more than 20 years ago, rituximab has dramatically changed the treatment landscape for patients with CD20+ lymphoid malignancies and has been included in the WHO model list of essential medicines [109]. As a result of its impressive efficacy, but also high price, it remained the highest grossing anti-cancer therapeutic through to 2016 [3]. With the development of next-generation mAbs, subcutaneous rituximab, and biosimilars, clinicians (and payers) will have more options to consider. With respect to next-generation molecules, the proposed advantages of the frontrunner obinutuzumab over rituximab will need to be weighed against the higher toxicity profile, IV-only formulation, and greater cost. Switching to a biosimilar for many practicing around the globe may provide a practical option, particularly for those practicing in developing countries, where the current cost of cancer treatment often exceeds the average per capita income by many multiples [110]. As further data emerges, it will be important for clinicians to be informed with respect to the potential advantages, risks, and cost-effectiveness of available CD20 mAbs, such that they can optimize treatment selection.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Nadler LM, Ritz J, Hardy R, Pesando JM, Schlossman SF, Stashenko P. A unique cell surface antigen identifying lymphoid malignancies of B cell origin. J Clin Invest. 1981;67(1):134–40.
Stashenko P, et al. Characterization of a human B lymphocyte-specific antigen. J Immunol. 1980;125(4):1678–85.
Pierpont TM, Limper CB, Richards KL. Past, present, and future of rituximab-the world’s first oncology monoclonal antibody therapy. Front Oncol. 2018;8:163.
Beers SA, Chan CHT, French RR, Cragg MS, Glennie MJ. CD20 as a target for therapeutic type I and II monoclonal antibodies. Semin Hematol. 2010;47(2):107–14.
Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–7.
Nadler LM, et al. Serotherapy of a patient with a monoclonal antibody directed against a human lymphoma-associated antigen. Cancer Res. 1980;40(9):3147–54.
Reff ME. The discovery of rituxan. In: The process of new drug discovery and development. 2nd ed. New York: Informa Healthcare; 2006, ed. C.G.O.D. Smith, J. 2006, New York: Informa Healthcare.
Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci U S A. 1984;81(21):6851–5.
Pyzik M, et al. FcRn: The architect behind the immune and non-immune functions of IgG and albumin. J Immunol (Baltimore, Md. : 1950). 2015;194(10):4595–603.
•• Salles G, et al. Rituximab in b-cell hematologic malignancies: a review of 20 years of clinical experience. Adv Ther. 2017;34(10):2232–73. Comprehensive review of rituximab across all malignant indications written by those involved in the pivotal trials and with extensive experience with this agent.
Kimby E. Tolerability and safety of rituximab (MabThera). Cancer Treat Rev. 2005;31(6):456–73.
• Davies A, et al. Subcutaneous rituximab for the treatment of b-cell hematologic malignancies: a review of the scientific rationale and clinical development. Adv Ther. 2017;34(10):2210–31. Overview of the development of the subcutaneous product.
Shpilberg O, Jackisch C. Subcutaneous administration of rituximab (MabThera) and trastuzumab (Herceptin) using hyaluronidase. Br J Cancer. 2013;109(6):1556–61.
Dakhil S, Hermann R, Schreeder MT, Gregory SA, Monte M, Windsor KS, et al. Phase III safety study of rituximab administered as a 90-minute infusion in patients with previously untreated diffuse large B-cell and follicular lymphoma. Leukemia & Lymphoma. 2014;55(10):2335–40.
Salar A, et al. Comparison of subcutaneous versus intravenous administration of rituximab as maintenance treatment for follicular lymphoma: results from a two-stage. J Clin Oncol: Phase IB Study; 2014.
Assouline S, Buccheri V, Delmer A, Gaidano G, McIntyre C, Brewster M, et al. Pharmacokinetics and safety of subcutaneous rituximab plus fludarabine and cyclophosphamide for patients with chronic lymphocytic leukaemia. Br J Clin Pharmacol. 2015;80(5):1001–9.
Davies A, Merli F, Mihaljević B, Mercadal S, Siritanaratkul N, Solal-Céligny P, et al. Efficacy and safety of subcutaneous rituximab versus intravenous rituximab for first-line treatment of follicular lymphoma (SABRINA): a randomised, open-label, phase 3 trial. Lancet Haematol. 2017;4(6):e272–82.
Lugtenburg P, Avivi I, Berenschot H, Ilhan O, Marolleau JP, Nagler A, et al. Efficacy and safety of subcutaneous and intravenous rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in first-line diffuse large B-cell lymphoma: the randomized MabEase study. Haematologica. 2017;102(11):1913–22.
Assouline S, Buccheri V, Delmer A, Gaidano G, Trneny M, Berthillon N, et al. Pharmacokinetics, safety, and efficacy of subcutaneous versus intravenous rituximab plus chemotherapy as treatment for chronic lymphocytic leukaemia (SAWYER): a phase 1b, open-label, randomised controlled non-inferiority trial. Lancet Haematol. 2016;3(3):e128–38.
Panizo C, et al. Safety of subcutaneous administration of rituximab during the first-line treatment of patients with non-Hodgkin lymphoma: the MabRella study. Blood. 2016;128(22):2971.
Rummel M, et al. Prefmab: final analysis of patient preference for subcutaneous versus intravenous rituximab in previously untreated CD20+ diffuse large B-cell lymphoma and follicular lymphoma. Blood. 2015;126(23):3972.
Rule S, Collins GP, Samanta K. Subcutaneous vs intravenous rituximab in patients with non-Hodgkin lymphoma: a time and motion study in the United Kingdom. J Med Econ. 2014;17(7):459–68.
Ponzetti C, et al. Potential resource and cost saving analysis of subcutaneous versus intravenous administration for rituximab in non-Hodgkin’s lymphoma and for trastuzumab in breast cancer in 17 Italian hospitals based on a systematic survey. Clinicoecon Outcomes Res. 2016;8:227–33.
De Cock E, et al. Time savings with rituximab subcutaneous injection versus rituximab intravenous infusion: a time and motion study in eight countries. PLoS One. 2016;11(6):e0157957.
Fargier E, Ranchon F, Huot L, Guerre P, Safar V, Dony A, et al. SMABcare study: subcutaneous monoclonal antibody in cancer care: cost-consequence analysis of subcutaneous rituximab in patients with follicular lymphoma. Ann Hematol. 2018;97(1):123–31.
Wolfromm A, et al. home administration of subcutaneous rituximab is safe and associated with significant cost saving: a single center experience. Blood. 2017;130(Suppl 1):4676.
Cragg MS, et al. The biology of CD20 and its potential as a target for mAb therapy. Curr Dir Autoimmun. 2005;8:140–74.
Johnson P, Glennie M. The mechanisms of action of rituximab in the elimination of tumor cells. Semin Oncol. 2003;30(1 Suppl 2):3–8.
Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. 2007;44(16):3823–37.
Chan HT, et al. CD20-induced lymphoma cell death is independent of both caspases and its redistribution into triton X-100 insoluble membrane rafts. Cancer Res. 2003;63(17):5480–9.
Cragg MS, et al. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood. 2003;101(3):1045–52.
Teeling JL, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 2004;104(6):1793–800.
Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2004;103(7):2738–43.
Alduaij W, Ivanov A, Honeychurch J, Cheadle EJ, Potluri S, Lim SH, et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood. 2011;117(17):4519–29.
Ivanov A, et al. Monoclonal antibodies directed to CD20 and HLA-DR can elicit homotypic adhesion followed by lysosome-mediated cell death in human lymphoma and leukemia cells. J Clin Invest. 2009;119(8):2143–59.
Golay J, et al. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000;95(12):3900–8.
Herter S, Herting F, Mundigl O, Waldhauer I, Weinzierl T, Fauti T, et al. Preclinical activity of the type II CD20 antibody GA101 (obinutuzumab) compared with rituximab and ofatumumab in vitro and in xenograft models. Mol Cancer Ther. 2013;12(10):2031–42.
Byrd JC, et al. The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction. Blood. 2002;99(3):1038–43.
Honeychurch J, Alduaij W, Azizyan M, Cheadle EJ, Pelicano H, Ivanov A, et al. Antibody-induced nonapoptotic cell death in human lymphoma and leukemia cells is mediated through a novel reactive oxygen species-dependent pathway. Blood. 2012;119(15):3523–33.
Di Gaetano N, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171(3):1581–7.
Vega MI, Huerta-Yepez S, Jazirehi AR, Garban H, Bonavida B. Rituximab (chimeric anti-CD20) sensitizes B-NHL cell lines to Fas-induced apoptosis. Oncogene. 2005;24(55):8114–27.
Bologna L, Gotti E, Manganini M, Rambaldi A, Intermesoli T, Introna M, et al. Mechanism of action of type II, glycoengineered, anti-CD20 monoclonal antibody GA101 in B-chronic lymphocytic leukemia whole blood assays in comparison with rituximab and alemtuzumab. J Immunol. 2011;186(6):3762–9.
Golay J, da Roit F, Bologna L, Ferrara C, Leusen JH, Rambaldi A, et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood. 2013;122(20):3482–91.
Mozessohn L, et al. Rituximab resistant follicular lymphoma: predictors of rituximab resistance, incidence of transformation and prognosis. Blood. 2011;118(21):4981.
Bonavida B. Postulated mechanisms of resistance of B-cell non-Hodgkin lymphoma to rituximab treatment regimens: strategies to overcome resistance. Semin Oncol. 2014;41(5):667–77.
Freeman CL, Sehn LH. A tale of two antibodies: obinutuzumab versus rituximab. Br J Haematol. 2018;182(1):29–45.
Rezvani AR, Maloney DG. Rituximab resistance. Best Pract Res Clin Haematol. 2011;24(2):203–16.
• Cartron G, Watier H. Obinutuzumab: what is there to learn from clinical trials? Blood. 2017;130(5):581–9. Very clear and comprehensive evaluation of this agent.
Keating MJ, Dritselis A, Yasothan U, Kirkpatrick P. Ofatumumab. Nat Rev Drug Discov. 2010;9(2):101–2.
Teeling JL, Mackus WJM, Wiegman LJJM, van den Brakel JHN, Beers SA, French RR, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol. 2006;177(1):362–71.
Wierda WG, Kipps TJ, Mayer J, Stilgenbauer S, Williams CD, Hellmann A, et al. Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol. 2010;28(10):1749–55.
Montillo M, Byrd JC, Hillmen P, O’Brien S, Barrientos JC, Reddy NM, et al. Long-term efficacy and safety in the resonate study: IBRUTINIB in patients with previously treated chronic lymphocytic leukemia (CLL) with up to four years follow-up. Hematol Oncol. 2017;35(S2):235–6.
Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–23.
Hillmen P, Robak T, Janssens A, Babu KG, Kloczko J, Grosicki S, et al. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet. 2015;385(9980):1873–83.
van Oers MH, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370–9.
Reyes C, et al. Cost-effectiveness analysis of obinutuzumab versus ofatumumab for previously untreated chronic lymphocytic leukemia (CLL). Blood. 2014;124(21):1324.
Wu Y, Wang Y, Gu Y, Xia J, Kong X, Qian Q, et al. Safety and efficacy of ofatumumab in chronic lymphocytic leukemia: a systematic review and meta-analysis. Hematology. 2017;22(10):578–84.
Marshall MJE, Stopforth RJ, Cragg MS. Therapeutic antibodies: what have we learnt from targeting CD20 and where are we going? Front Immunol. 2017;8:1245.
•• Sopp J, Cragg MS. Deleting malignant b cells with second-generation anti-cd20 antibodies. J Clin Oncol. 2018;36(22):2323–5. Brief and succint overview of the mechanism of action of rituximab compared to next generation antiCD20 Mabs.
Imhoff GWv, et al. Ofatumumab versus rituximab salvage chemoimmunotherapy in relapsed or refractory diffuse large b-cell lymphoma: the ORCHARRD study. J Clin Oncol. 2017;35(5):544–51.
Maloney DG, Fukuhara N., Ogura M, Larouche JF, Tournilhac O, Coleman M, Hong X, Fennessy Cota M, Woessner M, Tobinai K. A phase III study of ofatumumab vs rituximab in indolent B-cell non-Hodgkin lymphoma relapsed after rituximab-containing therapy (HOMER): results of the interim analysis in 21st Annual Congress of the European Hematology Association. 2016: Copenhagen, Denmar.
Mössner E, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell–mediated B-cell cytotoxicity. Blood. 2010;115(22):4393–402.
Niederfellner G, Lammens A, Mundigl O, Georges GJ, Schaefer W, Schwaiger M, et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood. 2011;118(2):358–67.
Konitzer JD, et al. Reformatting rituximab into human IgG2 and IgG4 isotypes dramatically improves apoptosis induction in vitro. PLoS One. 2015;10(12):e0145633.
Umana P, et al. GA101, a novel humanized type II CD20 antibody with glycoengineered Fc and enhanced cell death induction, exhibits superior anti-tumor efficacy and superior tissue B cell depletion in vivo. Blood. 2007;110(11):2348.
Salles G, Morschhauser F, Lamy T, Milpied N, Thieblemont C, Tilly H, et al. Phase 1 study results of the type II glycoengineered humanized anti-CD20 monoclonal antibody obinutuzumab (GA101) in B-cell lymphoma patients. Blood. 2012;119(22):5126–32.
Salles GA, Morschhauser F, Solal-Céligny P, Thieblemont C, Lamy T, Tilly H, et al. Obinutuzumab (GA101) in patients with relapsed/refractory indolent non-Hodgkin lymphoma: results from the phase II GAUGUIN study. J Clin Oncol. 2013;31(23):2920–6.
Sehn LH, Assouline SE, Stewart DA, Mangel J, Gascoyne RD, Fine G, et al. A phase 1 study of obinutuzumab induction followed by 2 years of maintenance in patients with relapsed CD20-positive B-cell malignancies. Blood. 2012;119(22):5118–25.
Sehn LH, Goy A, Offner FC, Martinelli G, Caballero MD, Gadeberg O, et al. Randomized phase II trial comparing obinutuzumab (GA101) with rituximab in patients with relapsed CD20+ indolent B-cell non-Hodgkin lymphoma: Final analysis of the GAUSS study. J Clin Oncol. 2015;33(30):3467–74.
Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370:1101–10.
Goede V, Fischer K., Dyer MJS, Müller L, Smolej L, Di Bernardo MC, Knapp A, Nielsen T, Hallek M Overall survival benefit of obinutuzumab over rituximab when combined with chlorambucil in patients with chronic lymphocytic leukemia and comorbidities: final survival analysis of the CLL11 study. in European Hematology Association 2018. EHA Learning Center.
Hiddemann W, Barbui AM, Canales MA, Cannell PK, Collins GP, Dürig J, et al. Immunochemotherapy with obinutuzumab or rituximab for previously untreated follicular lymphoma in the GALLIUM study: influence of chemotherapy on efficacy and safety. J Clin Oncol. 2018;36(23):2395–404.
Marcus R, Davies A, Ando K, Klapper W, Opat S, Owen C, et al. Obinutuzumab for the first-line treatment of follicular lymphoma. N Engl J Med. 2017;377(14):1331–44.
Friedberg JW. Progress in advanced-stage follicular lymphoma. J Clin Oncol. 2018;36(23):2363–5.
Casasnovas R-O, et al. Obinutuzumab versus rituximab in combination with ACVBP-14 or CHOP-14 following a PET-driven strategy in Aa-IPI 1-3 DLBCL patients (< 60 years): third planned interim and final analyses of the Gained trial. Blood. 2017;130(Suppl 1):190.
Vitolo U, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol. 2017;0(0):JCO.2017.73.3402.
Cartron G, Hourcade-Potelleret F, Morschhauser F, Salles G, Wenger M, Truppel-Hartmann A, et al. Rationale for optimal obinutuzumab/GA101 dosing regimen in B-cell non-Hodgkin lymphoma. Haematologica. 2016;101(2):226–34.
Rafiq S, Butchar JP, Cheney C, Mo X, Trotta R, Caligiuri M, et al. Comparative assessment of clinically utilized CD20-directed antibodies in chronic lymphocytic leukemia cells reveals divergent NK cell, monocyte, and macrophage properties. J Immunol. 2013;190(6):2702–11.
Vitolo U, et al. Obinutuzumab or rituximab plus CHOP in patients with previously untreated diffuse large B-cell lymphoma: final results from an open-label, randomized phase 3 study (GOYA). Blood. 2016;128(22):470.
Sacco JJ, Botten J, Macbeth F, Bagust A, Clark P. The average body surface area of adult cancer patients in the UK: a multicentre retrospective study. PLoS One. 2010;5(1):e8933.
Pott C, et al. Minimal residual disease in patients with follicular lymphoma treated with obinutuzumab or rituximab as first-line induction immunochemotherapy and maintenance in the phase 3 GALLIUM study. Blood. 2016;128(22):613.
Fischer K, al-Sawaf O, Fink AM, Dixon M, Bahlo J, Warburton S, et al. Venetoclax and obinutuzumab in chronic lymphocytic leukemia. Blood. 2017;129:2702–5.
Cramer P, von Tresckow J, Bahlo J, Robrecht S, Langerbeins P, al-Sawaf O, et al. Bendamustine followed by obinutuzumab and venetoclax in chronic lymphocytic leukaemia (CLL2-BAG): primary endpoint analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19(9):1215–28.
Launonen A, et al. Early disease progression predicts poorer survival in patients with follicular lymphoma (FL) in the GALLIUM study. Blood. 2017;130(Suppl 1):1490.
Casulo C, Byrtek M, Dawson KL, Zhou X, Farber CM, Flowers CR, et al. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the national LymphoCare study. J Clin Oncol. 2015;33(23):2516–22.
Oestergaard MZ, et al. Superiority of obinutuzumab over rituximab in a new molecular follicular lymphoma-like subgroup of DLBCL: results from an exploratory analysis of the phase 3 GOYA trial. Blood. 2017;130(Suppl 1):1543.
de Romeuf C, Dutertre CA, le Garff-Tavernier M, Fournier N, Gaucher C, Glacet A, et al. Chronic lymphocytic leukaemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcgammaRIIIA/CD16. Br J Haematol. 2008;140(6):635–43.
Miller J, et al. Ublituximab (TGTX-1101), a novel anti-CD20 monoclonal antibody (mAb), demonstrates activity in rituximab-sensitive and rituximab–resistant B non-Hodgkin lymphoma (B-NHL) pre-clinical <em>in vitro</em> and <em>in vivo</em> models. Blood. 2012;120(21):2756.
Le Garff-Tavernier M, et al. Analysis of CD16+CD56dim NK cells from CLL patients: evidence supporting a therapeutic strategy with optimized anti-CD20 monoclonal antibodies. Leukemia. 2011;25(1):101–9.
Sawas A, Farber CM, Schreeder MT, Khalil MY, Mahadevan D, Deng C, et al. A phase 1/2 trial of ublituximab, a novel anti-CD20 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma or chronic lymphocytic leukaemia previously exposed to rituximab. Br J Haematol. 2017;177(2):243–53.
Cazin B, et al. Multicentre phase I study with an 8-dose regimen of single agent anti-CD20 monoclonal antibody LFB-R603 in patients with relapsed chronic lymphocytic leukemia (CLL). Blood. 2011;118(21):2862.
Nastoupil LJ, et al. Tolerability and activity of chemo-free triplet combination of TGR-1202, ublituximab, and ibrutinib in patients with advanced CLL and NHL. J Clin Oncol. 2017;35(15_suppl):7511.
Prasad V, De Jesus K, Mailankody S. The high price of anticancer drugs: origins, implications, barriers, solutions. Nat Rev Clin Oncol. 2017;14(6):381–90.
S, G. Remarks from FDA Commissioner Scott Gottlieb, M.D., as prepared for delivery at the Brookings Institution on the release of the FDA’s Biosimilars Action Plan. 2018; Available from: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm613881.htm.
The Lancet H. Biosimilars: an optimistic outlook, but vigilance is needed. Lancet Haematol. 2017;4(8):e341.
Rioufol C, Salles G. Biosimilar monoclonal antibodies in lymphoma: a critical appraisal. Expert Rev Anticancer Ther. 2015;15(5):569–78.
Biosimilars in the EU - information guide for healthcare professionals. 2017 [cited 2018 2 SEPT]; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Leaflet/2017/05/WC500226648.pdf.
•• Vital EM, Kay J, Emery P. Rituximab biosimilars. Expert Opin Biol Ther. 2013;13(7):1049–62. Clear and comprehensive review of the manufacture, regulatory considerations, and overall development of rituximab biosimilars.
Schiestl M, Stangler T, Torella C, Čepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29:310–2.
Jurczak W, Moreira I, Kanakasetty GB, Munhoz E, Echeveste MA, Giri P, et al. Rituximab biosimilar and reference rituximab in patients with previously untreated advanced follicular lymphoma (ASSIST-FL): primary results from a confirmatory phase 3, double-blind, randomised, controlled study. Lancet Haematol. 2017;4(8):e350–61.
Association E.M. Summary of product characteristics: rixathon 2018; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003903/WC500232460.pdf.
Association E.M. Summary of product characteristics: truxima 2018; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004112/WC500222694.pdf.
Qureshi ZP, et al. Rituximab and biosimilars – equivalence and reciprocity. Biosimilars. 2013;2013(3):19–25.
Viswabandya A, et al. Pharmacokinetic and pharmacodynamic evaluation of a biosimilar rituximab in newly diagnosed diffuse large B-cell lymphoma (DLBCL) treated with R-CHOP (rituximab, cyclophosphamide, adriamycin, vincristine, prednisolone). Blood. 2007;110(11):4491.
Roy PS, et al. Comparison of the efficacy and safety of rituximab (Mabthera) and its biosimilar (Reditux) in diffuse large B-cell lymphoma patients treated with chemo-immunotherapy: a retrospective analysis. Indian J Med Paediatr Oncol. 2013;34(4):292–8.
Gota V, Karanam A, Rath S, Yadav A, Tembhare P, Subramanian P, et al. Population pharmacokinetics of Reditux, a biosimilar rituximab, in diffuse large B-cell lymphoma. Cancer Chemother Pharmacol. 2016;78(2):353–9.
Menon H, et al. Pharmacokinetic and pharmacodynamic properties of a biosimilar rituximab (Reditux<sup>®</sup>) are identical to the innovator brand MabThera<sup>®</sup>− experience from a tertiary cancer centre in Western India. Blood. 2014;124(21):2246.
Gulácsi L, Brodszky V, Baji P, Rencz F, Péntek M. The rituximab biosimilar CT-P10 in rheumatology and cancer: a budget impact analysis in 28 European countries. Adv Ther. 2017;34(5):1128–44.
Organization, W.H. WHO model list of essential medicines: 20th edition. 2017 [cited 2018 2 sept]; Available from: http://apps.who.int/iris/bitstream/handle/10665/273826/EML-20-eng.pdf?ua=1.
Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Global Oncol. 2017;3(5):596–610.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ciara L. Freeman has received research funding through grants from Roche and Genentech, has served on advisory boards for AbbVie and Celgene, and has received honoraria for service as a consultant from Seattle Genetics.
Laurie Sehn has received research funding through grants from Roche and Genentech and has received honoraria from Roche, Genentech, Seattle Genetics, AbbVie, Celgene, Amgen, Gilead, TG Therapeutics, Pfizer, Karyopharm, Apobiologix, Lundbeck, and Janssen for service as a consultant.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Lymphomas
Rights and permissions
About this article

Cite this article
Freeman, C.L., Sehn, L. Anti-CD20 Directed Therapy of B Cell Lymphomas: Are New Agents Really Better?. Curr Oncol Rep 20, 103 (2018). https://doi.org/10.1007/s11912-018-0748-0
Published:
DOI: https://doi.org/10.1007/s11912-018-0748-0