
Abstract
Purpose of Review
The purpose of this paper is to identify commonly used tyrosine kinase inhibitors (TKIs) that are associated with hypertension, primarily, vascular endothelial growth factor (VEGF) signaling pathway (VSP) inhibitors. We review the incidence, mechanism, and strategies for management of TKI-induced HTN. We hope to provide clinicians with guidance on how to manage similar clinical scenarios.
Recent Findings
Many of the newer VSP inhibitors are reviewed here, including cediranib, axitinib, pazopanib, and ponatinib. Trials utilizing prophylactic treatment with angiotensin system inhibitors (ASIs) are discussed as well as recent data showing an improvement in overall survival and progression-free survival in patients on ASIs and TKI-induced hypertension.
Summary
The incidence of TKI-induced HTN among the VEGF inhibitors ranges from 5 to 80% and is dose dependent. Newer generation small-molecule TKIs has a lower incidence. The mechanism of action involves VSP inhibition, leading to decreased nitric oxide and increased endothelin production, which causes vasoconstriction, capillary rarefaction, and hypertension. ASIs and calcium channel blockers are first-line therapy for treatment and are associated with improved overall survival. Nitrates and beta-blockers are associated with in vitro cancer regression; however, there is a paucity of trials regarding their use as an anti-hypertensive agent in the TKI-induced HTN patient population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The recent 2017 AHA/ACC guidelines define hypertension (HTN) as a systolic blood pressure (SBP) over 130 mmHg and a diastolic blood pressure (DBP) over 80 mmHg based on two or more measurements on two separate visits [1]. In an observational study of > 1 million adults ≥ 30 years old, elevated SBP and DBP were associated with increased risk of cardiovascular disease (CVD) incidence and angina, myocardial infarction, heart failure, stroke, peripheral arterial disease, and abdominal aortic aneurysm [2]. In the USA, HTN is the leading modifiable risk factor that causes CVD death [3].
With a prevalence of 37%, HTN is also the most common CVD comorbidity in cancer registries [4]. The etiology of HTN is vast and novel cancer therapies, such as tyrosine kinase inhibitors (TKIs) are associated with HTN. In fact, many physicians suggest that TKI-induced HTN is a surrogate for drug efficacy against tumor cells and the 2017 AHA/ACC guidelines specifically cite this class of drugs as a precipitant of HTN [1]. The TKIs most notorious for causing HTN are drugs that target the vascular endothelial growth factor (VEGF) signaling pathway (VSP). Both small-molecule and antibody VEGF inhibitors are well-tolerated first-line agents that have been shown to increase overall survival in a multitude of different cancers. VEGF inhibition primarily affects angiogenesis of tumor cells leading to tumor death; however, inhibition of angiogenesis can also result in HTN through endothelial-dependent pathways, alterations in capillary density, and even micro-emboli.
While guidelines recommend treatment of HTN for long-term mortality risk reduction, most patients that receive VSP inhibitor therapy receive the drug for a short period and have limited life expectancy, calling into question the need to treat their HTN. However, serious adverse events, discontinuation of therapy, and decrease in overall survival can occur with untreated TKI-induced HTN. New data suggests that treating TKI-induced HTN can lead to improved overall survival as well as cancer progression-free survival due to control of a modifiable CVD risk factor and the ability to stay on life-saving cancer therapy for longer periods [5]. Besides HTN, VSP inhibition can lead to other serious adverse cardiac events such as arterial thrombotic disease, cardiomyopathy, and ischemic heart disease with varying incidence; however, discussion of these adverse events are beyond the scope of this article. The incidence, mechanism, and management of TKI-induced HTN via VSP inhibition will be reviewed here.
Incidence
HTN in earlier studies using TKIs was defined most commonly by using the Common Terminology Criteria for Adverse Events (CTCAE) versions 2.0 or 3.0 as described in Table 1 [6]. Some studies also used the Joint National Committee of Prevention, Detection, Evaluation, and Treatment of High Blood Pressure 7 (JNC 7) guidelines that defined HTN as BP ≥ 140/90. The CTCAE version 4.0 was modified to parallel the JNC 7 guidelines in an effort to achieve more consistency. The recent change in HTN guidelines published by the ACC/AHA in 2017 is thus not reflected in these studies.
TKIs have been associated with increased incidence of HTN but the incidence can vary depending on the drug, pre-existing conditions, dose, and type of tumor. Some of the risk factors include pre-existing HTN with SBP ≥ 160 mmHg or DBP ≥ 100 mmHg, diabetes, established cardiac or renal disease, older age, cigarette smoking, dyslipidemia, family history of premature CVD, obesity, and elevated fasting plasma glucose levels [7]. The incidence of HTN for some of the commonly used TKIs are discussed below and summarized in Table 2.
Bevacizumab
Bevacizumab is a monoclonal antibody that binds VEGF and has been used to treat colon cancer, lung cancer, glioblastoma, renal cell carcinoma (RCC), and age-related macular degeneration. It has been studied in several trials with an overall incidence of HTN from 4 to 35%. Grade 3 HTN developed in 11 to 18%. Severe HTN requiring discontinuation of therapy or hospitalization was noted in 1.7% of patients treated with bevacizumab [8]. HTN can occur with drug initiation within the first year of treatment. A median interval from first dose of bevacizumab to the onset of HTN was 131 days (range of 7–316 days) as reported by Yang et al. [9].
Studies have also shown a possible association between increased risk of developing HTN with dose and tumor type. In a meta-analysis of 19 randomized control trials done by An et al. that included 12,949 cancer patients with various solid tumors, the relative risk ratios of significantly elevated BP in patients receiving 5 and 2.5 mg/kg per week were 7.17 (95% CI, 3.91–13.13) and 4.11 (95% CI 2.49–6.78) respectively. It was also noted that the risk of developing HTN was higher in the patients with RCC (RR 13.77, 95% CI 2.28–83.15) and breast cancer patients (RR 18.83, 95% CI 1.23–292.29) who received bevacizumab at 5 mg/kg per week [10].
Sunitinib
Sunitinib is a TKI used more commonly in the treatment of metastatic RCC and gastrointestinal stromal tumor (GIST). The incidence of HTN in patients treated with sunitinib has ranged from 5 to 24% in various trials [11]. In a meta-analysis done by Zhu et al., a total of 4999 patients with RCC and other malignancies from 13 clinical trials were included for analysis. The incidence of all-grade and high-grade HTN in patients treated with sunitinib were 21.6% (95% CI 18.7–24.8%) and 6.8% (95% CI 5.3–8.8%) respectively. There was an increased risk of high-grade HTN (RR = 22.72, 95% CI 4.48 to 115.29, p < 0.001) noted when compared to controls. There was also a significant difference detected between RCC and non-RCC in terms of all-grade HTN (RR 1.32, 95% CI 1.18–1.48%, p < 0.001) and high-grade HTN (RR 1.57, 95% CI 1.22–2.02%, p = 0.001) which may in part be related to renal dysfunction caused by sunitinib [12]. Azizi et al. showed that BP levels may increase as early as a week after initiating treatment with sunitinib [13].
Sorafenib
Sorafenib is a multi-kinase inhibitor and has several targets including the VEGF-receptor. It is most commonly used for RCC, hepatocellular carcinoma, and advanced thyroid carcinoma. The incidence of HTN in various trials has ranged from 7 to 43% for sorafenib [11]. In a meta-analysis done by Wu et al. that included 4599 patients in nine studies with RCC or other solid tumors, the overall incidence of all-grade HTN was 23.4% (95% CI 16.0–32.9%) and 5.7% (2.5–12.6%) for high-grade HTN (grade 3 or 4). The RR for increased risk of all-grade HTN was 6.11 (2.4415.32, p < 0.001) compared with control subjects. No significant difference was detected in the incidence of sorafenib-associated HTN between patient with RCC and non-RCC cancer [14]. Maitland et al. noted BP elevations from sorafenib as early as within 1 day and more readily detected when steady state of sorafenib reached in a week [15].
Pazopanib
The overall incidence of HTN in patients who underwent therapy with pazopanib is around 42% in various trials [11]. It is most commonly used for advanced RCC or soft tissue sarcoma. In a small retrospective study of 35 patients with metastatic RCC at a single medical center, Pinkhas et al. noted an incidence of pazopanib-induced HTN in 57% of patients. New-onset HTN was noted in 43% of patients. The overall median time to development of pazopanib-induced HTN was 24.5 days. A baseline SBP > 130 mmHg was associated with a higher risk of developing HTN with pazopanib [16].
Axitinib
The overall incidence of HTN in patients treated with axitinib was 40% [11]. It is most commonly used for treatment of RCC as well. A recent analysis by Qin et al. looked at 26 trials, including 4790 patients diagnosed with RCC and treated with axitinib or sorafenib monotherapy and noted a higher incidence of HTN (24.9 vs. 7.9%) in patients receiving axitinib versus sorafenib [17].
Cediranib
Cediranib is an oral small-molecule TKI of VEGF-receptor 1 (VEGFR-1), VEGFR-2, VEGFR-3 among others, and is being explored in the treatment of ovarian, non-small cell lung cancer, glioblastoma, and colon cancer. A phase II trial done by Robinson and colleagues of 46 women, 31 women (67%) developed HTN by day 3; 87% developed HTN by end of study and 43% developed higher grade HTN (grade 3 or more) [18].
Ponatinib
Ponatinib is a third-generation TKI that is a potent chemotherapeutic agent used most often against resistant cases of chronic myeloid leukemia (CML), especially in patients with the BCR-ABL1T3151 mutation [19]. Unlike other BCR-ABL TKIs, ponatinib has a significant inhibitory effect on VEGFR-2 similar to molecules specifically designed to inhibit VSP [19]; therefore, it is no surprise that there is a significant association with HTN. Overall incidence of various degrees of HTN in various studies was as high as 68% [11]. A dose-dependent cardiovascular toxicity is noted with ponatinib as well as less tolerance of higher doses in older patient with history of diabetes or ischemic events [19].
Of note, ponatinib trials showed a significant incidence of arterial thrombotic events at 12 months, as high as 14%. Patients with traditional cardiovascular risk factors had a greater predisposition to these adverse outcomes. Due to continuing high incidence at 24 months and similar trends in subsequent studies, there was transient suspension of the drug in the US market and a restricted FDA use label [19].
Dasatinib
Dasatinib is a second-generation TKI approved for front-line therapy of CML based on superior results over imatinib [20]. Initial safety reviews isolated pleural effusions as a common therapy side effect and source of shortness of breath (SOB) [21]. At 36-month follow-up in the same study, however, there was a 3% incidence pulmonary arterial hypertension (PAH), which increased to 5% at 5 years [22]. The findings triggered an FDA warning for evaluation of signs and symptoms of cardiopulmonary disease in patients before and during dasatinib treatment, with notation that pleural effusion and anemia were still the most common etiology of SOB symptoms [19].
VEGF Therapeutic Mechanism of Action
Aberrant activation of receptor tyrosine kinases (RTKs) are implicated in multiple cancers making them an ideal therapeutic target. With the advent of VEFG inhibitors, oncologists can employ another targeted pathway to battle cancer with more manageable adverse effects. Currently, these antibodies and small molecules are utilized as first-line treatment, and beyond, in the metastatic setting with improvement in overall survival and progression-free survival. These agents are very well tolerated and have demonstrated an improvement in quality of life.
Human RTKs are cell surface receptors that span the cell membrane consisting of an extracellular domain along with an intracellular tyrosine kinase domain. There are 58 human RTKs which are further sub classified into 20 families and function as key regulators of critical cellular function [23]. The extracellular portion has binding sites for growth factors, which are endogenous molecules that promote cell proliferation and induce neovascularization [24, 25]. In general, growth factor binding activates the receptor by inducing structural changes to the extracellular domain and causing a cascade of signals downstream that activate the intracellular domain via phosphorylation [24, 25].
There are three primary growth factors that may bind to the RTKs and the ensuing effects on the cell are dependent on the type of growth factor. Vascular endothelial growth factor (VEGF) is the dominant growth factor in controlling angiogenesis. Epidermal growth factor (EGF) is responsible for differentiation and apoptosis. Platelet-derived growth factor (PDGF) plays a role in cell growth, cell division, and angiogenesis [26].
VEGF-A is the dominant growth factor that mediates angiogenesis by binding to VEGFR on the endothelial cells stimulating cell proliferation, migration, and survival [27]. VEGFRs are expressed on both normal endothelium and tumor cells. There are both anti-VEGF antibodies as well as anti-VEGFR antibodies that target the cell surface receptor for VEGF and also block VEGF signaling. Both types of antibodies are currently utilized as targets for cancer therapy [28, 29].
The upregulation of both VEGF-A mRNA and VEGFR has been demonstrated in majority of human tumor cells. In order for malignant cells to progress and metastasize, the tumor must recruit its own blood supply by increasing angiogenesis and forming blood vessels into avascular tumor masses [30]. VEGF inhibition by anti-VEGF antibodies such as bevacizumab exert their action by neutralizing VEGF, causing regression of existing blood vessels, and depleting the tumor vascular supply causing the cells to die. There is evidence that the remaining blood vessels are then normalized which allows traditional chemotherapy delivery to tumor cells. Chronic VEGF inhibition by bevacizumab also inhibits new and recurrent blood vessel growth [31, 32].
VEGF-Induced Hypertension Mechanism of Action
Regression of blood vessels by VEGF signaling leads to tumor death, but also has untoward consequences on the normal vasculature: HTN, proteinuria, and arterial thromboemboli.
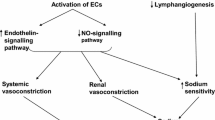
VEGF signaling normally enhances endothelial-derived nitric oxide (NO) production, which then acts on smooth muscle cells to vasodilate (Fig. 1) [6]. VSP inhibitors interfere with NO production in the arteriolar walls which then leads to vasoconstriction, increased vascular resistance, and ultimately, HTN [33]. In mice studies done by Facemire and colleagues, an anti-VEGFR2 antibody caused a rapid and sustained increase in BP of ≈10 mmHg. The HTN in response to the anti-VEGFR2 antibody was associated with significant reductions in the expression of endothelial and neuronal NO synthases in the kidney [34]. These findings were replicated in Bevacizumab trials [35].

Pathophysiology of hypertension induced by inhibition of vascular endothelial growth factor (VEGF) signaling pathway inhibition. Proposed mechanisms leading to vascular endothelial growth factor (VEGF) signaling pathway inhibitor-induced hypertension. a Activation of VEGF-receptor 2 by VEGF-A leads to subsequent activation of multiple pathways including phosphatidylinositol-3-kinase (PI3K)-AKT. PI3K-AKT phosphorylates and activates endothelial nitric oxide synthase (eNOS), increasing NO production. NO migrates to adjacent vascular smooth muscle cells and binds soluble guanylate cyclase (sGC), leading to cGMP generation and subsequent vasodilation mediated by cGMP-dependent kinases. When VEGF signaling pathway is inhibited, NO pathway is suppressed and endothelin (ET)-1 pathway is stimulated, promoting vasoconstriction and subsequent hypertension. The source of ET-1 is unknown. b VEGF maintains capillary network integrity. When VEGF signaling pathway is inhibited, rarefaction or reduction of the density of capillary beds, may occur; however, this is likely a minor contributor to antiangiogenic therapy-induced hypertension. From de Jesus-Gonzalez N, Robinson E, Moslehi J, Humphreys BD. Management of antiangiogenic therapy-induced hypertension. Hypertension, Vol. 60, Issue 3, pages 607–15, accessible at http://hyper.ahajournals.org/content/60/3/607. Reproduced with kind permission from Wolters Kluwer Health, Inc.
Normal VEGF signaling mediates endothelial homeostasis. VSP inhibition leads to endothelial dysfunction, stimulating the release of Endothelin-1 (ET-1), a potent vasoconstrictor that may also play a role in mediating HTN [36]. Kappers et al. have reported a parallel rise in ET-1 and HTN in both human and rodents treated with sunitinib [37]. Regorafenib has been shown to drastically increase ET-1 levels, while co-administration of an endothelin receptor antagonist blunts the hypertensive response triggered by other anti-angiogenic therapies [38, 39]. While the distinct pathway linking ET-1 and VEGF inhibition continues to be studied, ET-1 has been suggested to act in a paracrine or autocrine manner [37].
When VSP is inhibited, NO is suppressed and ET-1 is stimulated, promoting vasoconstriction and subsequent hypertension. Another aspect of endothelial dysfunction that has a minor role in the development of hypertension has to do with micro-capillary rarefaction. VEGF maintains capillary network integrity, thus when inhibition occurs, rarefaction or reduction of the density of capillary beds can occur. Mouse models reveal a 30% regression of tracheal mucosal capillary networks by 21 days when treated with VEGF inhibitors, which then reverses with anti-angiogenic therapy discontinuation [40]. In patients with metastatic colorectal carcinoma receiving bevacizumab, the capillary density of the dorsum of the finger was reduced by 10% after 6 months of treatment, which was associated with increase in BP [41].
Subsets of patients who develop anti-angiogenic therapy-induced HTN also develop proteinuria and micro-angiopathic hemolysis, which can lead to acute kidney injury. Glomerular endotheliosis represents a specific variant of thrombotic micro-angiopathy that is characterized by glomerular endothelial swelling with loss of endothelial fenestrae and occlusion of the capillary lumens. Associated thrombosis is unusual. Recent evidence suggests that this unusual glomerular lesion is mediated by a soluble VEGFR that deprives glomerular endothelial cells of the VEGF that they require, leading to cellular injury and disruption of the filtration apparatus with subsequent proteinuria [42].
In a study by Patel et el, seven patients developed a preeclampsia-like syndrome characterized by HTN and proteinuria after starting therapy with TKI, identified clinically after developing edema, HTN, proteinuria, and/or hypoalbuminemia [43]. A possible mechanism of the renal effects is the inhibition of glomerular VEGFR signaling by TKI. Bevacizumab is associated with a dose-dependent risk of proteinuria and HTN [44]. The effects of bevacizumab and other VEGF inhibitors are thought to be from delayed development of glomerular endotheliosis and slow rise of BP, similar to the pathophysiology of preeclampsia in pregnancy.
Management
The incidence of TKI-induced HTN may actually be a biomarker for the drug’s efficacy in the treatment of cancer. In a meta-analysis of metastatic RCC patients receiving sunitinib, sunitinib-induced HTN was associated with improved overall survival and progression-free survival; however, there was a small but statistically significant increase in adverse renal events [45••]. Based on these findings, close monitoring and treatment of HTN is important to prevent serious adverse events that are known to occur with unmanaged HTN in patients on VSP inhibitors [46, 47]. Acceptable BP control would allow patients to receive the most effective dose of TKI, remain on therapy for the duration of their oncologic treatment, and improve their overall survivorship [48].
The National Cancer Institute (NCI) recommends identification of those patients at highest risk for TKI-induced HTN with a pre-treatment CVD health assessment [7]. This includes a detailed history and physical where concomitant clinical risk factors and subclinical organ damage are identified. Characteristics such as age, smoking and second-hand smoke exposure history, sleep apnea, psychological stress, family history of HTN, chronic kidney disease, unhealthy diet, obesity, diabetes, and hyperlipidemia will help identify those patients at greater risk of developing HTN during course of therapy. Additional measurements of left ventricular hypertrophy on 2D echocardiogram, carotid wall thickness, electrocardiogram, serum creatinine, 24-h urine protein, fasting blood glucose, and lipid panel may help characterize severity of comorbidities prior to initiation of therapy and help guide medical management [1, 7].
Patients with one or more clinical CVD risk factors are considered high risk and, per NCI clinical trial protocols, should have weekly BP monitoring with first cycle of VSP inhibitor and then every 2–3 weeks for duration of treatment [7]. This corresponds with clinical trials that suggest onset of HTN occurs after first or second doses of VSP inhibitors [49]. During this time, BP assessment should be performed in office. If elevated, repeat assessment and elevation is required during a second visit for diagnosis of TKI-induced HTN. For patients with white coat HTN (SBP ≥ 120 mmHg and ≤ 160 mmHg), home BP recordings can be used to support or negate the diagnosis. Reversible causes of HTN, such as poor pain control and high sodium diet, should be ruled out and appropriately managed [7].
Because the primary goal in the oncology setting is to prevent HTN related adverse events, BP should be reduced to a goal of < 140/90 mmHg (130/80 mmHg in chronic kidney disease and diabetic patients), as recommended by the Cardiovascular Toxicities Panel of the NCI in 2010 [7]. Stringent BP control is not recommended and may lead to less-effective VSP inhibitor dosing and/or polypharmacy [49]. If BP goal is not achieved, VSP inhibitor therapy can still be initiated given there is improvement in BP and values are below those associated with adverse cardiac events [7]. Finally, once TKI therapy is completed, patients need adjustments in their anti-hypertensive regimens to prevent hypotension.
Pharmacotherapy
Agents available for treatment of TKI-induced HTN are the same as for treatment of essential HTN. The proposed mechanism of action of TKI-induced HTN as well as clinical experience guide specific drug selection and are reviewed below. A summary of commonly used anti-hypertensive drugs, as discussed below, are provided in Table 3.
It has been suggested that angiotensin system inhibitors (ASIs) (angiotensin converting enzyme inhibitors (ACE-Is) and angiotensin receptor blockers (ARBs)) may be the preferred anti-hypertensive agent in the management of TKI-induced HTN, due to their beneficial effects on plasminogen activator inhibitor-1 expression and proteinuria [35]. ASIs also increase the release of endothelial NO, the production of which is reduced by treatment with VSP inhibition as discussed above [7]. Blood pressure lowering is relatively rapid producing changes within 24–48 h. Most importantly, ASIs have been shown to improve overall survival and progression-free survival. Izzedine et al. conducted a retrospective study on 213 patients receiving sunitinib and/or ASIs for metastatic RCC and associated pre- and therapy-related HTN. Through multivariate COX analysis, they found that patients who received the drug before or within one cycle of sunitinib, had a significant increase in overall survival (median, 26.4 months; p < 0.0001), and in progression-free survival (median, 8.1 months; p < 0.0001) than patients who were not on ASIs [5]. The authors hypothesize whether these findings are due to interactions between the ASI pathway and cancer proliferation or if HTN serves as a biomarker of drug efficacy. Similar findings were observed in a large meta-analysis of 4736 patients with metastatic RCC who underwent therapy with a small-molecule TKI. McKay et al. showed that ASIs were associated with improved survival ( [50••]). It is our practice to use this drug as first-line therapy, given the patient does not have stage 4 or 5 chronic kidney disease.
Due to their primary vasodilatory mechanism, dihydropyridine calcium channel blockers are attractive agents in the management of TKI-induced HTN particularly in patients with severe HTN requiring multi-drug therapy. In the study reported by Mir et al., amlodipine 5 mg daily resulted in control of HTN in 80–90% of patients who developed de novo or worsening HTN while on bevacizumab for metastatic non-small cell lung cancer, colorectal cancer, and ovarian cancer [51]. Unfortunately, the use of nifedipine had come into question due to early studies showing nifedipine-induced VEGF-secretion [52]. However, rat models evaluating cediranib in colorectal cancer showed nifedipine was more effective than captopril in reducing diastolic BP without affecting the anti-tumor activity [53]. Due to anti-hypertensive effectiveness with dihydropyridine CCBs, a trial on prophylactic use of CCBs in patients receiving cediranib was conducted and found to be effective for the management of HTN; this has influenced all ongoing cediranib trials to include similar protocols [54].
Non-dihyhdropyridine CCBs such as verapamil and diltiazem are CYP3A4 inhibitors, which may lead to drug-drug interactions with chemotherapeutic agents metabolized by the cytochrome P450 pathway such as sunitinib and sorafenib. Bevacizumab is metabolized via a different pathway and therefore these drugs may be considered [55].
Nitrate therapy might be considered attractive given the effect of VSP on nitric oxide production in the mechanism of inducing HTN. A 3-patient series of TKI-induced resistant HTN showed that short-acting isosorbide dinitrate effectively controlled blood pressure when three other medications would not, including ASI and dihydropyridine [56]. There is, however, data that suggests nitrate therapy may compromise anti-angiogenic benefits since VSP-induced angiogenesis is dependent on downstream NO production [57].
Although thiazide diuretics are standard therapy in the treatment of essential hypertension, they may be less effective than calcium channel blockers or ASIs in treating VSP-induced hypertension [49]. However, if there is limited success with many of the agents discussed above, thiazide diuretics, such as chlorthalidone and hydrochlorothiazide, should be considered.
There is little clinical data looking at beta-blocker therapy in VSP inhibition-induced HTN. Mouse models suggest nebivolol stimulates NO production, which induces an endothelial dependent vasodilation and reduction in blood pressure [58]. Additional mouse models have been able to demonstrate an anti-tumor effect with beta-blocker use [59]. Future studies should look at beta-blockers in the VSP inhibitor-induced HTN population for its effectiveness as an anti-hypertensive and its anti-cancer properties. It is still an ideal agent to use in patients with known coronary artery disease and/or chest pain.
Based on the elevated circulating levels of ET-1 observed in clinical and experimental studies with TKI-induced HTN, endothelin receptor blockade may be considered to mitigate the rise in BP [60]. The side effects of salt and water retention and peripheral edema likely will limit the applicability of these drugs to the treatment of HTN. Additionally, endothelin receptor antagonists are currently indicated only for the treatment of pulmonary hypertension. One final consideration for management of TKI-induced HTN is discontinuation or decrease in dose of the offending TKI. This is usually reserved for refractory HTN, hypertensive crises, or severe HTN (e.g., proteinuria and acute kidney injury). After discontinuation, it is recommended to use a different VSP inhibitor when re-challenging the patient [55].
Conclusion
In summary, aberrant RTK function is implicated in a multitude of cancers making it an ideal therapeutic target. TKIs target many receptors to varying degrees, but it is a specific drug’s affinity for the VEGFR that drives its association with HTN. The incidence of some degree of clinically significant HTN in clinical trials ranges from 5 to 80%; however, the “real life” incidence is likely higher given many trials may exclude patients with difficult-to-control HTN. The pathophysiologic mechanism driving the HTN is likely complex and multifactorial. A decrease in endothelial dependent production in nitric oxide, an increase in circulating ET-1, and a decrease in capillary density all likely play a role in increased peripheral vasoconstriction, which then results in HTN. Based on the mechanism of action, nitrate therapies and ET-1 antagonists seem like likely therapeutic agents; however, only case reports exist demonstrating their benefit. The most robust data exists for ASIs and CCBs, which allow for improved endothelial function and result in vasodilation and drop in BP. It is the practice of this writing group to use them as first-line agents. ASIs in particular are nephro-protective and have been associated with improved overall survival. Non-dihydropyridine CCBs are avoided due to potential drug-drug interactions. Use of anti-hypertensives is theorized to allow patients to tolerate higher effective doses of their TKIs, prevent disruption of therapy, and prevent serious adverse events from TKIs. Prospective randomized clinical trials are needed to test these theories in the future, which would provide valuable data to allow the most effective, life-saving, management in this patient population.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Whelton PK, Carey RM, Aronow WS et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2017.
Rapsomaniki E, Timmis A, George J, Pujades-Rodriguez M, Shah AD, Denaxas S, et al. Blood pressure and incidence of twelve cardiovascular diseases: lifetime risks, healthy life-years lost, and age-specific associations in 125 million people. Lancet. 2014;383:1899–911.
Danaei G, Ding EL, Mozaffarian D, Taylor B, Rehm J, Murray CJL, et al. The preventable causes of death in the United States: comparative risk assessment of dietary, lifestyle, and metabolic risk factors. PLoS Med. 2009;6:e1000058.
Jain M, Townsend RR. Chemotherapy agents and hypertension: a focus on angiogenesis blockade. Curr Hypertens Rep. 2007;9:320–8.
Izzedine H, Derosa L, Le Teuff G, Albiges L, Escudier B. Hypertension and angiotensin system inhibitors: impact on outcome in sunitinib-treated patients for metastatic renal cell carcinoma. Ann Oncol. 2015;26:1128–33.
Common Terminology Criteria for Adverse Events (CTCAE) V 3.0. .
Maitland ML, Bakris GL, Black HR, Chen HX, Durand JB, Elliott WJ, et al. Initial assessment, surveillance, and management of blood pressure in patients receiving vascular endothelial growth factor signaling pathway inhibitors. J Natl Cancer Inst. 2010;102:596–604.
Yeh ET, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis and management. J Am Coll Cardiol. 2009;53:2231–47.
Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody for metastatic renal cancer. N Engl J Med. 2003;349:427–34.
An MM, Zou Z, Shen H, Liu P, Chen ML, Cao YB, et al. Incidence and risk of significantly raised blood pressure in cancer patients treated with bevacizumab: an updated meta-analysis. Eur J Clin Pharmacol. 2010;66:813–21.
Chang HM, Okwuosa TM, Scarabelli T, Moudgil R, Yeh ETH. Cardiovascular complications of Cancer therapy: best practices in diagnosis, prevention, and management: part 2. J Am Coll Cardiol. 2017;70:2552–65.
Zhu X, Stergiopoulos K, Wu S. Risk of hypertension and renal dysfunction with an angiogenesis inhibitor sunitinib: systematic review and meta-analysis. Acta Oncol. 2009;48:9–17.
Azizi M, Chedid A, Oudard S. Home blood-pressure monitoring in patients receiving sunitinib. N Engl J Med. 2008;358:95–7.
Wu S, Chen JJ, Kudelka A, Lu J, Zhu X. Incidence and risk of hypertension with sorafenib in patients with cancer: a systematic review and meta-analysis. Lancet Oncol. 2008;9:117–23.
Maitland ML, Kasza KE, Karrison T, Moshier K, Sit L, Black HR, et al. Ambulatory monitoring detects sorafenib-induced blood pressure elevations on the first day of treatment. Clin Cancer Res. 2009;15:6250–7.
Pinkhas DHTSS. Assessment of pazopanib-related hypertension, cardiac dysfunction and identification of clinical risk factors for their development. Cardio-Oncology. 2017;3
Qin F, Yu H, Xu CR, Chen HH, Bai JL. Safety of axitinib and sorafenib monotherapy for patients with renal cell carcinoma: a meta-analysis. J Biomed Res. 2018;32:30–8.
Robinson ES, Matulonis UA, Ivy P, Berlin ST, Tyburski K, Penson RT, et al. Rapid development of hypertension and proteinuria with cediranib, an oral vascular endothelial growth factor receptor inhibitor. Clin J Am Soc Nephrol. 2010;5:477–83.
Moslehi JJ, Deininger M. Tyrosine kinase inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol. 2015;33:4210–8.
Kantarjian HM, Shah NP, Cortes JE, Baccarani M, Agarwal MB, Undurraga MS, et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood. 2012;119:1123–9.
Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–41.
Jabbour E, Kantarjian HM, Saglio G, Steegmann JL, Shah NP, Boque C, et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood. 2014;123:494–500.
Gale NW, Yancopoulos GD. Growth factors acting via endothelial cell-specific receptor tyrosine kinases: VEGFs, angiopoietins, and ephrins in vascular development. Genes Dev. 1999;13:1055–66.
Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604–17.
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–31.
Veikkola T, Alitalo K. VEGFs, receptors and angiogenesis. Semin Cancer Biol. 1999;9:211–20.
Kuwai T, Kitadai Y, Tanaka S, Onogawa S, Matsutani N, Kaio E, et al. Expression of hypoxia-inducible factor-1alpha is associated with tumor vascularization in human colorectal carcinoma. Int J Cancer. 2003;105:176–81.
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76.
Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–27.
Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, Lieberman G, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21:60–5.
Verheul HM, Lolkema MP, Qian DZ, et al. Platelets take up the monoclonal antibody bevacizumab. Clin Cancer Res. 2007;13:5341–7.
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62.
Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer. 2007;96:1788–95.
Facemire CS, Nixon AB, Griffiths R, Hurwitz H, Coffman TM. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54:652–8.
Dincer M, Altundag K. Angiotensin-converting enzyme inhibitors for bevacizumab-induced hypertension. Ann Pharmacother. 2006;40:2278–9.
Bohm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res. 2007;76:8–18.
Kappers MH, van Esch JH, Sluiter W, Sleijfer S, Danser AH, van den Meiracker AH. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. 2010;56:675–81.
de Jesus-Gonzalez N, Robinson E, Penchev R, von Mehren M, Heinrich MC, Tap W, et al. Regorafenib induces rapid and reversible changes in plasma nitric oxide and endothelin-1. Am J Hypertens. 2012;25:1118–23.
Kappers MH, de Beer VJ, Zhou Z, et al. Sunitinib-induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension. 2012;59:151–7.
Baffert F, Le T, Sennino B, et al. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Phys Heart Circ Phys. 2006;290:H547–59.
Mourad JJ, des Guetz G, Debbabi H, Levy BI. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann Oncol. 2008;19:927–34.
Stillman IE, Karumanchi SA. The glomerular injury of preeclampsia. J Am Soc Nephrol. 2007;18:2281–4.
Patel TV, Morgan JA, Demetri GD, George S, Maki RG, Quigley M, et al. A preeclampsia-like syndrome characterized by reversible hypertension and proteinuria induced by the multitargeted kinase inhibitors sunitinib and sorafenib. J Natl Cancer Inst. 2008;100:282–4.
Zhu X, Wu S, Dahut WL, Parikh CR. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: systematic review and meta-analysis. Am J Kidney Dis. 2007;49:186–93.
•• Rini BI, Cohen DP, Lu DR, et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst. 2011;103:763–73. This article is the largest study to highlight that hypertension can be used as a biomarker for VSP inhibition success in treating a cancer. It lends to further research questions of treatment success, failures, and possible relation to dosing.
Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370:2011–9.
Schmidinger M, Zielinski CC, Vogl UM, Bojic A, Bojic M, Schukro C, et al. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008;26:5204–12.
Piccirillo JF, Tierney RM, Costas I, Grove L, Spitznagel EL Jr. Prognostic importance of comorbidity in a hospital-based cancer registry. Jama. 2004;291:2441–7.
de Jesus-Gonzalez N, Robinson E, Moslehi J, Humphreys BD. Management of antiangiogenic therapy-induced hypertension. Hypertension. 2012;60:607–15.
•• McKay RR, Rodriguez GE, Lin X, et al. Angiotensin system inhibitors and survival outcomes in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2015;21:2471–9. Largest meta-analysis that shows an association with use of a specific anti-hypertensive medication and improvement in overall survival during therapy with a VSP inhibitor. Validates the importance of treatment of hypertension and modifies how future trials of TKI inhibitors may be run.
Mir O, Coriat R, Ropert S, Cabanes L, Blanchet B, Camps S, et al. Treatment of bevacizumab-induced hypertension by amlodipine. Investig New Drugs. 2012;30:702–7.
Miura S, Fujino M, Matsuo Y, Tanigawa H, Saku K. Nifedipine-induced vascular endothelial growth factor secretion from coronary smooth muscle cells promotes endothelial tube formation via the kinase insert domain-containing receptor/fetal liver kinase-1/NO pathway. Hypertens Res. 2005;28:147–53.
Curwen JO, Musgrove HL, Kendrew J, Richmond GH, Ogilvie DJ, Wedge SR. Inhibition of vascular endothelial growth factor-a signaling induces hypertension: examining the effect of cediranib (recentin; AZD2171) treatment on blood pressure in rat and the use of concomitant antihypertensive therapy. Clin Cancer Res. 2008;14:3124–31.
Langenberg MH, van Herpen CM, De Bono J, et al. Effective strategies for management of hypertension after vascular endothelial growth factor signaling inhibition therapy: results from a phase II randomized, factorial, double-blind study of Cediranib in patients with advanced solid tumors. J Clin Oncol. 2009;27:6152–9.
Nazer B, Humphreys BD, Moslehi J. Effects of novel angiogenesis inhibitors for the treatment of cancer on the cardiovascular system: focus on hypertension. Circulation. 2011;124:1687–91.
Dirix LY, Maes H, Sweldens C. Treatment of arterial hypertension (AHT) associated with angiogenesis inhibitors. Ann Oncol. 2007;18:1121–2.
Cooke JP, Losordo DW. Nitric oxide and angiogenesis. Circulation. 2002;105:2133–5.
Mason RP, Jacob RF, Corbalan JJ, Szczesny D, Matysiak K, Malinski T. The favorable kinetics and balance of nebivolol-stimulated nitric oxide and peroxynitrite release in human endothelial cells. BMC Pharmacol Toxicol. 2013;14:48.
Pasquier E, Street J, Pouchy C, Carre M, Gifford AJ, Murray J, et al. Beta-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br J Cancer. 2013;108:2485–94.
Lankhorst S, Kappers MH, van Esch JH, Danser AH, van den Meiracker AH. Mechanism of hypertension and proteinuria during angiogenesis inhibition: evolving role of endothelin-1. J Hypertens. 2013;31:444–54. discussion 454
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Megha Agarwal, Nidhi Thareja, Melody Benjamin, Andre Akhondi, and George D. Mitchell declare they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Cardio-oncology
Rights and permissions
About this article

Cite this article
Agarwal, M., Thareja, N., Benjamin, M. et al. Tyrosine Kinase Inhibitor-Induced Hypertension. Curr Oncol Rep 20, 65 (2018). https://doi.org/10.1007/s11912-018-0708-8
Published:
DOI: https://doi.org/10.1007/s11912-018-0708-8