
Abstract
In recent years, a revolution in the management of chronic lymphocytic leukemia (CLL) has centered on the targeting of the B cell receptor (BCR) signaling pathway. Our improved understanding of the biology of cell signaling in CLL and the development of oral kinase inhibitors directed at the BCR pathway has led to the approval of two new agents and has the potential to radically change the treatment of CLL in both the relapsed/refractory and upfront settings. In this review, we will describe the underlying biology of the BCR signaling pathway. We will discuss the landmark clinical trials resulting in the approval of the Bruton tyrosine kinase (BTK) inhibitor ibrutinib and the PI3Kδ inhibitor idelalisib. We will highlight ongoing trials that are evaluating the use of combinations of these agents with standard chemotherapy. We will evaluate some of the emerging data regarding toxicity, potential off-target effects, and mechanisms of resistance to BCR signaling pathway blockade. Finally, we will highlight some of the next-generation BCR pathway inhibitors currently in development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
For many patients, CLL is a disease of slow accumulation [1]. Patients progress through a somewhat reliable course as defined many years ago by the Rai staging system [2]. Initial, isolated lymphocytosis progresses into lymphadenopathy with eventual splenomegaly and cytopenias. Traditionally, patients were initiated on therapy only when their disease burden generated symptoms or when symptoms were imminent. Therapy with standard chemo-immunotherapeutic regimens, including rituximab-bendamustine (BR) and rituximab-cyclophosphamide-fludarabine (FCR), resulted in significant clinical responses for the majority of patients [3,4,5,6,7]. However, the above regimens have toxicity and, for the most part, patients would eventually relapse. For a subset of patients with high-risk disease as indicated by the presence of an unmutated IGHV gene or the presence of high-risk cytogenetic features (17p deletion and 11q deletion), responses to traditional chemo-immunotherapy have the potential to be less pronounced or more short lived [8, 9]. Patients with mutated IGHV respond particularly well to treatment with FCR [10]. In a recent update of the CLL 8 trial, comparing FCR to fludarabine and cyclophosphamide, median survival following treatment with FCR has not been met and overall survival at 5 years is 86.3%. A second experience with FCR noted progression-free survival (PFS) of 30.9% at a median follow-up of 12.8 years [11•]. In the population of patients with mutated IGHV, the PFS at the same time point was 53.9%. In addition, a plateau was seen on the PFS curve in patients with mutated IGHV, with no patients relapsing after 10.4 years, suggesting the potential of cure.
A better understanding of the underlying biology of the BCR signaling pathway and its importance in CLL survival suggested the possibility of targeted therapy for CLL.
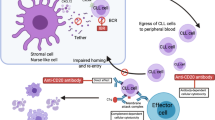
BCR pathway signaling is activated through antigenic stimulation of the extracellular domain of the BCR (Fig. 1). Upon activation of the BCR, CD79a and CD79b are recruited, with activation of spleen tyrosine kinase (SYK) and LYN kinase. SYK and LYN kinases phosphorylate immunoreceptor tyrosine-based activation motifs (ITAMs) leading to an activation cascade that activates BTK and PI3Kδ. BTK, once activated, phosphorylates and activates phospholipase C gamma 2 (PLCγ2) and through downstream mediators, results in the release of intracellular calcium stores, the propagation of the BCR signal, and upregulation of transcription factors such as nuclear factor κB (NF-κB) [12, 13]. The ultimate result of this signaling pathway is the activation of a number of cellular processes including integrin activation, chemokine-mediated migration, and proliferation of B cells. The above process in normal, healthy B cells allows for humoral response to antigens recognized by the BCR. In CLL, there appears to be dysregulated signaling through the BCR pathway resulting in improved tumor survival and proliferation [14, 15]. While there do not appear to be direct activating mutations of the BCR pathway in CLL, it remains unclear whether the inappropriate BCR signaling in CLL is a result of the recognition of an antigen by the BCR or whether there is non-antigen-dependent BCR pathway activity [16, 17]. In contrast, activating mutations have been identified in the BCR signaling pathway in patients with diffuse large B cell lymphoma (DLBCL). These mutations, including mutations of the ITAM signaling modules of CD79a and CD79b, are frequently seen in activated B cell (ABC) DLBCL but rarely in germinal center B cell DLBCL [18]. In DLBCL, these mutations associate with response to BTK inhibitors, but most if not all CLLs appear to be dependent on this pathway even without mutations. Given this dependence of CLL on the BCR signaling pathway, a number of small-molecule inhibitors of kinases within the pathway have been explored as therapeutic agents in CLL.

BCR pathway signaling is activated through antigenic stimulation of the extracellular domain of the BCR with recruitment of CD79a and CD79b and activation SYK and LYN, leading to a downstream activation cascade that activates BTK and PI3Kδ and ultimately the upregulation of transcription factors, such as nuclear factor κB (NF-κB). The result of this signaling pathway is the activation of a number of cellular processes including integrin activation, chemokine-mediated migration, and proliferation of B cells. Idelalisib and Ibrutinib are orally available inhibitors of PI3Kδ and BTK, respectively, resulting in interruption of this pathway
The Road to Approval
There are currently two approved agents for the management of CLL that target the BCR signaling pathway. Ibrutinib, an inhibitor of BTK, has been approved for use in patients with CLL and in patients with CLL with a 17p deletion. In addition, ibrutinib has been approved for treatment of patients with mantle cell lymphoma, Waldenstrom’s macroglobulinemia, and marginal zone lymphoma. Idelalisib has been approved in combination with rituximab for patients with CLL for whom rituximab would be considered appropriate therapy due to other comorbidities, and for patients with small lymphocytic lymphoma (SLL) who have received at least two prior systemic therapies. Idelalisib is also indicated for patients with relapsed follicular B cell non-Hodgkin lymphoma who have received at least two prior systemic therapies.
Ibrutinib
Ibrutinib irreversibly binds to the activation site of the BTK enzyme at the cysteine-481 amino acid [19]. Preclinical studies demonstrated that treatment with ibrutinib led to decreased CLL proliferation and tumor migration. More recent data suggest that treatment with ibrutinib decreases the ability of CLL to utilize free fatty acids for growth and proliferation [20]. RNA sequencing on CD19+ tumor cells from patients with CLL treated with ibrutinib performed before therapy, 1 month and 6 months after starting treatment, demonstrated that 653 genes were differentially expressed on treatment compared to baseline, with more profound changes seen at the later time point [21]. Nine pathways were significantly inhibited by ibrutinib, including pathways involved in cytokine signaling, cell adhesion, systemic lupus, p53 response, MAPK signaling, cell cycle, focal adhesion, calcium signaling, and Wnt signaling.
An initial phase 1 study of ibrutinib demonstrated only moderate toxicity and significant efficacy [22•]. Fifty-six patients with a variety of B cell malignancies were treated over seven cohorts. The majority of adverse events were of grade 1 or 2. Grade 3 or 4 cytopenias included neutropenia (12.5%), thrombocytopenia (7.2%), and anemia (7.1%). Only two dose-limiting toxicities occurred. One patient with a known history of drug sensitivity had a grade 3 allergic reaction. One patient had a dose interruption of more than 7 days because of transient grade 2 neutropenia. Of the 50 patients evaluable for tumor response, 60% achieved an objective response. Eleven of 16 (69%) patients with CLL or SLL had responses. A CLL-specific phase 1b-2 study investigating the safety and efficacy of ibrutinib in 85 patients demonstrated a similar toxicity profile and a more dramatic response rate [23•]. Again, the majority of adverse events were grade 1 or 2 and included diarrhea, fatigue, and upper respiratory infection. Most adverse events resolved without suspension of therapy. Ten patients (12%) had a grade 2 or higher pneumonia. Infections were more common earlier in therapy. The overall response rate was 71%, and 15–20% of patients had a partial response with persistent lymphocytosis. RESONATE, a subsequent randomized phase III registration trial, demonstrated the superiority of ibrutinib to ofatumumab [24•, 25•]. Three hundred ninety-one patients with relapsed or refractory CLL were randomized 1:1 to receive ibrutinib or ofatumumab. At a median follow-up of 9.4 months, median PFS had not been reached with ibrutinib as compared to a median PFS of 8.1 months for treatment with ofatumumab (HR 0.22, p < 0.001). Ibrutinib also demonstrated an overall survival benefit, with a hazard ratio for death of 0.43 for treatment with ibrutinib (p = 0.005).
Ibrutinib has been investigated as an agent for patients with CLL harboring abnormalities in TP53. A single-agent, phase II trial enrolled 51 patients with relapsed or refractory disease that had abnormalities in TP53 [26]. Thirty-three previously untreated patients and 15 with relapsed or refractory disease were evaluable for response at 24 weeks. Ninety-seven percent of the untreated patients achieved an objective response. Eighty percent of the patients with relapsed or refractory disease responded. At 24 months, PFS was 82%. RESONATE 17, a single-agent, phase II registration study investigated the role of ibrutinib for patients with relapsed or refractory CLL with a 17p deletion [27]. One hundred forty-five patients were enrolled. Patients had a median age of 64 years, two prior therapies, and a median follow-up of 11.5 months. Eighty-three percent of patients had an overall response with 24 month PFS of 63%.
In the upfront setting, a small cohort of elderly patients was investigated as part of the original CLL-specific phase Ib/II trial [28]. Thirty-one patients, a minority with high-risk disease as defined by abnormalities in 17p (6%) or 11q (3%), were enrolled with a response rate of 77% and a 24-month PFS of 96.3%. In RESONATE-2, ibrutinib was compared to chlorambucil in a randomized, open-label trial in patients older than 65, with CLL without deletion 17p, and requiring initial therapy [29]. Although older, this patient population was quite healthy, with only 31–33% having an elevated comorbidity (CIRS) score >6. At a median follow-up of 18.4 months, treatment with ibrutinib resulted in a significant improvement in PFS relative to chlorambucil (not reached vs. 18.9 months) with a decrease in the risk of death or progression of 84%. In addition, overall survival at 24 months was significantly different, favoring ibrutinib (98 vs. 85%, p = 0.001). At the time of publication, 87% of patients in the ibrutinib arm continue on therapy. Updated efficacy was recently reported and notes that at a median time on study of 28.6 months, ibrutinib continues to have significant efficacy with 88% reduction in risk of progression or death compared to chlorambucil [30]. In addition, the quality of responses has improved over time with 18% of patients now achieving a CR. Furthermore, treatment-limiting adverse events have decreased in frequency with extended follow-up. Seventy-nine percent of this elderly patient population continues on therapy. Taken together, these studies represent the foundation for the approval of ibrutinib and lay the groundwork for future combination strategies.
Idelalisib
Idelalisib is an oral, bioavailable, small-molecule inhibitor of PI3K p110δ, an isoform expressed selectively in hematopoietic cells [31]. The initial efficacy of idelalisib in CLL was demonstrated in a phase I trial-treating patients with relapsed or refractory CLL [32•]. Fifty-four patients with adverse characteristics, including bulky lymphadenopathy, extensive prior therapy, refractory disease, unmutated IGHV, and deletion of 17p or TP53 mutations, were included. The most common grade 3 adverse events included pneumonia in 20% of patients and neutropenic fever in 11% of patients. Eighty-one percent of patients had a nodal response. Median PFS was 15.8 months but at the recommended phase 2 dose of 150 mg BID or higher, it was 32 months. A randomized phase III trial compared the combination of idelalisib and rituximab to rituximab plus placebo in patients with CLL with comorbid conditions that prevented treatment with standard chemotherapy regimens [33•]. Two hundred twenty patients with CLL with major coexisting illnesses were randomized to therapy. Overall response rate in the combination arm was significantly better than in the rituximab placebo arm (81 vs. 13%; odds ratio, 29–92%; p < 0.001). Median PFS was not met in the idelalisib and rituximab arm and was 5.5 months in the rituximab alone arm (hazard ratio for progression or death in the idelalisib group, 0.15; p < 0.001). Overall survival at 12 months was 92% in the combination arm and 80% in the rituximab arm (p = 0.02). Serious adverse events were similar between the two groups, 40% in combination therapy and 35% in patients receiving rituximab alone. A phase II study investigated the role of idelalisib in the treatment of non-Hodgkin lymphoma, including SLL, in patients who were refractory to rituximab and an alkylating agent [34]. The overall response rate was 58%, all of which were partial responses. The median survival for all patients was 20.3 months, and the overall survival at 1 year was estimated to be 80%.
Combination Studies
Based on the above success, many trials investigating the combinations of ibrutinib and idelalisib with standard chemo-immunotherapy and other targeted agents are currently enrolling or were recently reported.
Ibrutinib was combined with the chemo-immunotherapeutic regimens BR and FCR as treatment for patients with relapsed or refractory CLL in a multicenter phase Ib trial [35]. Thirty patients received BR-ibrutinib and three patients received FCR-ibrutinib. The overall response rate for BR-ibrutinib was 93.3%, and all three of the FCR-ibrutinib patients obtained a complete remission.
The Helios trial compared rituximab and bendamustine to the combination of ibrutinib, rituximab, and bendamustine in a randomized, double blind, placebo controlled trial [36]. Five hundred seventy-eight patients with CLL or SLL that had previously been treated were enrolled to therapy (289 in each group) with a primary endpoint of PFS. At a median follow-up of 17 months, PFS was significantly improved with ibrutinib, rituximab, and bendamustine combination therapy relative to the control arm (PFS not reached vs. 13.3 months, hazard ration 0.203, 95% CI 0.15–0.276; p < 0.0001). PFS at 18 months was 79% in the ibrutinib-BR arm and 24% in the BR arm (p < 0.0001). The most common grade 3 or higher adverse event was neutropenia, which was 54% in the ibrutinib BR arm and 51% in the BR arm. Thrombocytopenia was 15% in both arms.
Patients on the Helios, RESONATE, and RESONATE 2 trials were examined for the impact of deletion 11q on clinical outcomes [37]. No difference in response or overall survival was observed in patients receiving ibrutinib, even though 11q deletion is traditionally considered a marker of high-risk disease. In patients not receiving ibrutinib, the presence of deletion11q was associated with a shorter PFS.
Ibrutinib and rituximab were studied in a phase II single arm trial in patients with CLL with high-risk characteristics, as characterized by deletion 17p, deletion 11q, the presence of a TP53 mutation, or a PFS of less than 36 months from prior chemo-immunotherapy [38]. Forty patients were enrolled, 20 of whom had deletion 17p or TP53 mutations, 13 with deletion 11q, and seven with a short PFS. The overall response rate was 95%, and the 18-month PFS in all patients was 78%.
Ibrutinib has been combined with the PI3Kδ inhibitor TGR-1202 in a strategy that targets both BTK and PI3Kδ [39]. This phase Ib/II trial enrolled 17 patients with CLL and 11 patients with mantle cell lymphoma. The phase I portion of the trial was well tolerated with no dose-limiting toxicities. In the CLL population, the overall response rate was 82%. All patients with prior PI3K inhibitor exposure responded to therapy as did one of the two patients with prior ibrutinib exposure. The overall response rate in mantle cell lymphoma was 67%.
Given the efficacy and tolerability of ibrutinib FCR seen in the three patients treated on the above phase 1b study, a phase II trial is investigating ibrutinib in combination with FCR as upfront therapy [40]. Thirty-five patients, 65 years old and younger, were enrolled to therapy. Twenty-eight patients have undergone primary endpoint restaging. The overall response rate was 100%. Of the 26 patients for whom bone marrow biopsy results were available, the rate of CR with bone marrow negative for minimal residual disease is 39%, and 13 of the 17 patients with a partial response were also found to have no evidence of minimal residual disease on bone marrow biopsy. The overall rate of MRD negativity in the bone marrow at end of therapy was >80%. Hematologic and infectious toxicity was low, possibly related to the mandatory use of growth factors and infectious prophylaxis.
A phase Ib/II trial is investigating the combination of ibrutinib, obinutuzumab, and venetoclax in patients with relapsed and refractory CLL [41]. This non-chemotherapy-containing regimen targets CLL at the BCR pathway (ibrutinib), CD20 (obinutuzumab), and via inhibition of a mediator of apoptosis (venetoclax). Twelve patients have been treated in the phase Ib portion of the trial. Toxicities for the combination were consistent with those reported for the single agents, and no dose-limiting toxicities have been observed, such that each drug can be given at its full single-agent dose. Of the six patients evaluable for response, five have had a partial response and one patient has had a complete response.
Given the significant activity of idelalisib in combination with rituximab, its use in combination with a number of different agents has been investigated.
Idelalisib in combination with rituximab and bendamustine as compared to rituximab and bendamustine was investigated in a randomized, phase III, placebo-controlled, double blind trial, treating patients with relapsed or refractory CLL [42]. Four hundred sixteen patients were enrolled. At a median follow-up of 14 months, the median PFS was 20.8 months in the idelalisib-containing arm and 11.1 months in the placebo arm (p < 0.0001). An increased risk of grade 3 or higher infections was seen in the idelalisib-containing arm (39 vs. 25%). Treatment emergent deaths were seen in 11% of patients in the idelalisib-containing arm and 7% in the control arm. Updated efficacy data recently demonstrated that the combination of idelalisib BR had improved overall survival relative to BR (not reached vs. 41 months, p = 0.036) (43).
Beyond Response: Toxicity, Off-target Effects, and Resistance
Treatment-related Lymphocytosis
During the initial trials with ibrutinib and idelalisib, significant, transient lymphocytosis was noted following the initiation of therapy. The lymphocytosis appears to be mediated by the migration of CLL from the protected lymph node and bone marrow niches via alterations in CXCL12 and CXCR4. CLL that has migrated to the peripheral blood has shortened survival relative to disease in one of the protected niches, and this phenomenon contributes to the efficacy of BCR pathway inhibition [44, 45]. In reaction to this effect, the CLL response criteria have been revised to accept that lymphocytosis following BCR pathway inhibition is not diagnostic of resistant disease, and a new category of response for patients with a lymphocytosis and partial nodal response, PR-L, is now included [46,47,48].
Ibrutinib-specific Toxicity
BTK is important for signaling via the collagen receptor glycoprotein VI in platelets [49, 50]. Patients with X-linked agammaglobulinemia have a mutation in BTK resulting in a block in B cell development resulting in severe hypogammaglobulinemia and also difficulty with platelet aggregation in response to collagen. TEC is a kinase related to BTK, which is also inhibited by ibrutinib. TEC is involved in G protein-coupled receptor and integrin-mediated signaling in human blood platelets [51]. Bleeding has been a recurrent toxicity with ibrutinib therapy. In the initial phase I/II study in non-Hodgkin lymphoma, no clear increase in bleeding risk was seen [22•]. With increased enrollment on further studies, the increased rate of bleeding became clear. Based on the increased bleeding risk, patients receiving treatment with warfarin have been excluded from trials, and it is recommended to hold ibrutinib for 3–7 days prior to and following any surgery.
In the randomized trial comparing ibrutinib monotherapy to ofatumumab, a significant difference in bleeding-related adverse events of any grade was observed (44 vs. 12%) [24•]. However, major hemorrhage was reported in only two patients on ibrutinib and three patients receiving ofatumumab. In the upfront trial randomizing 269 patients to ibrutinib or chlorambucil, 4% of the patients in the ibrutinib group had a grade 3 or higher hemorrhage or central nervous system hemorrhage of any grade [29]. A retrospective, single-center study investigated the incidence of bleeding in patients treated with ibrutinib [52]. Seventy-one patients were available for analysis. Seventy percent of patients were noted to be treated with an antiplatelet agent, mostly aspirin, 7% of patients were treated with an anticoagulant, and 13% of patients were treated with combination antiplatelet and anticoagulant medications. In addition, any medications that interacted with cytochrome P450 3A4 (CYP3A4), which metabolizes ibrutinib, were recorded. Bleeding of any grade occurred in 56% of patients and major bleeding in 18% of patients. Of the nine patients receiving combined antiplatelet and anticoagulant therapy, 78% suffered a major bleeding event. Of the ten patients on ibrutinib not receiving antiplatelet, anticoagulation, or CYP3A4 interacting agents, no patients had a major bleeding event. This study highlights the real-world experience and risk of bleeding while on ibrutinib, as well as the need for caution when combining with anticoagulation. A second retrospective study examined bleeding events in 437 patients receiving ibrutinib [53]. 33.6% of patients were on aspirin. Major bleeding was seen in 14 patients (3.2%). Half of the bleeding events were seen in patients not receiving other antihemostatic medications. Approximately, one third of the patients who developed major bleeding resumed ibrutinib without a recurrent major bleeding event. Light transmission aggregometry on 23 patients receiving ibrutinib demonstrated reductions in collagen-mediated platelet aggregation with a significant association between the degree of inhibition and the occurrence of clinical bleeding or bruising [54]. The collagen defect reversed following cessation of ibrutinib therapy.
An additional Ibrutinib-specific toxicity that became clear in the phase III trial of ibrutinib vs. ofatumumab was the increased risk for the development of atrial fibrillation. Atrial fibrillation developed in ten patients receiving ibrutinib but only one patient receiving ofatumamab at the time of the first report of the RESONATE trial [24•]. The risk benefit-ratio of continued treatment for any patients receiving ibrutinib who develop atrial fibrillation must be considered. A recent analysis from four of the randomized trials of ibrutinib showed that the incidence was highest in the first 6 months but that new cases continued at a lower rate indefinitely. In this study, most patients were able to resume ibrutinib but the small subset with atrial fibrillation recurrence frequently had to stop ibrutinib [55]. A retrospective analysis of 56 patients who developed atrial fibrillation or atrial flutter while on ibrutinib found that 22 of the 56 patients stopped ibrutinib at the time of atrial fibrillation. Only three of the 22 patients who initially interrupted ibrutinib were able to successfully restart treatment. Of these 22 patients, seven (32%) died of progression or other complications. Those patients who interrupted ibrutinib at the time of atrial fibrillation onset had an inferior PFS relative to those patients who remained on ibrutinib (19 vs. 27 months, p = 0.023) [56], although no difference in OS was seen. This highlights the difference between the relatively healthy patient population treated on clinical trials relative to the real-world experience and points to the need for better data to manage atrial fibrillation secondary to ibrutinib.
Long-term follow-up of patients receiving ibrutinib on the RESONATE and RESONATE-2 studies summarized the rate of adverse events over time and includes up to 5 years of follow-up of 330 patients [57]. The majority of adverse events were grade 1 or 2. Grade 3 events included diarrhea (5%), arthralgia (2%), hypertension (7%), rash (4%), bleeding (6%, including two events of grade 4 bleeding and one grade 5 bleeding event), fatigue (3%), and atrial fibrillation (5%).
Patients on ibrutinib receiving antiplatelet or anticoagulant therapies for atrial fibrillation should be monitored for signs of bleeding. Atrial fibrillation should be managed appropriately and if it persists, consider the risks and benefits of ibrutinib therapy. As we gain more data and experience on the risks of bleeding and atrial fibrillation, formal recommendations may become more explicit. Currently, this decision is made on a per patient basis.
Ibrutinib Effects on Immunity
While ibrutinib is a potent inhibitor of Bruton tyrosine kinase, it also has significant binding to 19 other kinases with an IC50 less than 100 nM. One clinically significant off-target effect appears to be mediated by inhibition of the IL-2 inducible (ITK) kinase [58]. Studies using samples from CLL patients treated with ibrutinib and preclinical models have demonstrated that ibrutinib binds to and inhibits ITK resulting in a selective advantage for Th1-based immune responses relative to Th2 immunity. In addition, it has been shown that long-term treatment with ibrutinib results in improvement in CLL-mediated T cell immune suppression. At baseline, T cells from patients with CLL demonstrate minimal proliferation in response to stimulation with CD3/CD28 beads. After 5–11 cycles of ibrutinib, this proliferative defect is largely reversed, and T cell proliferation kinetics are similar to those of young healthy donors [59]. This improved proliferation resulted in improved ex vivo expansion of T cells generated for CAR-T infusion. This may be mediated by downregulation of PD1 and other markers of T cell exhaustion. In mouse models of mantle cell lymphoma, concurrent treatment with ibrutinib potentiated CAR T cell response and survival [60]. Treatment with ibrutinib prior to T cell leukapheresis appears to improve the CAR T phenotype, as measured by increased levels of immunoregulatory cytokines, though it may also increase the risk for toxicity mediated by cytokine release syndrome [61]. The effects of ibrutinib on T cells were compared to the effects of acalabrutinib, a BTK inhibitor which does not inhibit ITK [62]. Ibrutinib, but not acalabrutinib, was found to expand activated T cell populations. Both ibrutinib and acalabrutinib decreased the expression of PD-1 on T cells, most significantly on central memory T cells. Working with samples from patients on the HELIOS trial, treatment with ibrutinib has been found to result in a decrease in Th17 CD4+ cells, which have the potential to be autoimmune and protumorigenic, as well as a decrease in the percentage of Treg cells, with an overall increase in T cell activation [63]. Treatment with ibrutinib decreased serum levels of a number of inflammatory cytokines including IL6, IL8, IFN gamma, and TNF alpha. This may represent a reversal of the T cell pseudoexhaustion seen in CLL [64]. Ibrutinib inhibits rituximab-induced NK cell cytokine secretion in a dose-dependent manner resulting in decreased ADCC-mediated cytotoxicity [65]. This effect on ADCC appears to be mediated by inhibition of ITK, as inhibition of NK function was not seen following exposure to CGI1746, a BTK specific inhibitor. It also appears that ibrutnib can inhibit macrophage function [66]. These effects on ADCC and macrophage function could have implications for trials combining ibrutinib with agents that are dependent on ADCC, mostly antibodies, although to date, no evidence of antagonism in vivo has been observed.
The effects of ibrutinib therapy on response to pneumococcal conjugate vaccine PCV13 were investigated in a prospective study [67]. Four CLL patients receiving treatment with ibrutinib and four CLL patients not on therapy were administered PCV13. Zero of the four patients receiving ibrutinib generated a twofold or greater increase in three or more pneumococcal serotypes, while all four of the CLL control patients generated a response, suggesting that ibrutinib may have significant effects on humoral immunity. A mouse model of autoimmune hemolytic anemia was used to demonstrate the effects of BTK inhibition on antibody production [68]. Following treatment with BTK inhibition, mice were injected with rat red blood cells weekly for 4 weeks to induce an antibody response. Mice treated with acalabrutinib had significantly less autoantibody than mice treated with a control compound (p = 0.001), suggesting the possible use of BTK inhibition for the clinical management of autoimmune hemolytic anemia. A number of case reports demonstrate improvement in autoimmune hemolytic anemia in CLL patients treated with ibrutinib [69,70,71,72].
Idelalisib-specific Toxicity
Following the initial trials investigating the use of idelalisib in relapsed and refractory CLL, a series of upfront trials were terminated secondary to the observation of increased risk of death related to infection for patients randomized to combinations containing idelalisib. This experience was communicated to health care professionals via an FDA alert, and a black box warning is now included in the idelalisib product insert. (http://www.fda.gov/Drugs/DrugSafety/ucm490618.htm). The majority of the deaths were due to bacterial sepsis sometimes associated with neutropenia, but PJP and CMV infections were also seen, leading to the recommendation that patients receiving idelalisib should be on PJP prophylaxis and should have regular monitoring for the development of CMV infection.
In addition to the increased rate of death related to infection, increased likely autoimmune toxicity related to lymphocytic infiltrates was observed in the upfront setting. A phase II study investigating the combination of idelalisib and ofatumumab as upfront therapy for CLL enrolled 24 patients. Nineteen (79%) experienced a grade 1 or higher elevation in transaminases, and 13 subjects (54%) experience grade 3 or higher transaminitis [73]. The development of transaminitis occurred before the initiation of ofatumumab, at a median time of 28 days. Two patients with transaminitis had liver biopsies performed, which demonstrated an activated perforin-positive CD8-positive lymphocytic infiltrate. All episodes of transaminitis resolved with drug hold or the initiation of immunosuppression; the latter was required if idelalisib was to be restarted. This autoimmune hepatitis was associated with younger patient age, mutated immunoglobulin heavy chains, and reduced circulating Tregs after receiving idelalisib. Similar findings in relapsed patients with colitis on idelalisib have been reported. Twelve of 29 samples had an inflammatory and ischemic pattern, with a mixed appearance with both apoptotic and ischemic and inflammatory features. Five of the colon samples were found to have CMV involvement and one sample had HHV6 [74], but no viral infections were associated with the hepatitis.
One potential mechanism for the development of the hepatic lymphocytic infiltrate is the effects of PI3K inhibition on regulatory T cells (Tregs). PI3K activity has been shown to be critical to Treg development and function. Initial studies with PI3K deficient mice demonstrated decreased numbers of Tregs and decreased Treg function as demonstrated by their inability to protect against experimental colitis in adoptive transfer experiments [75]. Similar lack of Treg function was seen when PI3K deficiency was limited to T cells. Mice lacking PI3K function in their T cells develop a spontaneous Sjogren’s syndrome-like disease and have decreased numbers of peripheral Tregs [76]. Ex vivo investigation into the mechanism by which PI3K promotes Treg function has demonstrated that blockade of PI3K prevents IL-2, IL-4, IL-7, and IL-15 maintenance of regulatory function in Tregs [77]. It has been further shown that PI3K pathway inhibitors decrease Treg production of granzyme B, a mechanism by which Tregs selectively induce apoptosis in antigen-presenting B cells and effector T cells and a key mechanism for Treg-mediated suppression of tumor clearance [77]. Mice with an inactivating knock-in mutation in the p110δ isoform of PI3K have decreased Treg number and are more effective at clearing Leishmania major infections. Increased clearance of Leishmania was abrogated by the adoptive transfer of wild type Tregs [78]. Furthermore, Tregs from mice with a kinase-dead mutant p110δ PI3K have inferior suppressive capacity relative to wild type Tregs [79]. Given the increased risk of infection and risk of death related to infection coupled with significant idelalisib-mediated liver, colonic, and pulmonary injury, the use of idelalisib in CLL continues to be defined. Given the favorable toxicity profile of ibrutinib relative to idelalisib, clinical practice has favored ibrutinib in the majority of clinical settings. In patients with a high risk of bleeding, idelalisib may be the preferred agent. Idelalisib use after prior ibrutinib is also appropriate, although prospective data on its activity in these patients is lacking.
Resistance
While responses to ibrutinib are seen in the majority of patients, over time, patients relapse. One mechanism of resistance that has been identified is the development of mutations in the BCR signaling pathway that affect ibrutinib binding to BTK and downstream signaling [80]. Whole-exome sequencing of six patients with acquired resistance to ibrutinib identified a cysteine to serine mutation at the ibrutinib-binding site of BTK in five patients. Functional studies demonstrated that this mutation resulted in a decrease of the affinity of ibrutinib for BTK (dissociation constant, 0.2 vs. 10.4 nM). In addition to the mutations in BTK, three mutations in two patients were identified in PLCγ2. Both of these appeared to be gain of function mutations, resulting in autonomous BCR pathway activity. Baseline samples did not have the above mutations, consistent with treatment-related acquisition. Further analysis of the PLCγ2 mutations demonstrated BTK independent activation following BCR engagement, consistent with the development of a BTK bypass pathway [81]. Analyzing previously banked samples from 15 patients with a clinical relapse related to either a BTK or PLCγ2 mutation demonstrated that the resistant clone was able to be identified a median of 9.3 (95% CI 7.6–11.7) months prior to clinical relapse [82]. In vitro studies of Ramos cell lines exposed to ibrutinib developed resistance through BCL2 upregulation and interferon response genes [83]. In Namalwa cell lines conditioned by exposure to increasing doses of ibrutinib, BCR signaling pathway genes were overexpressed, suggesting constitutive activation of BTK and downstream kinases as a potential mechanism of resistance.
Given that BTK mutation is a common mechanism of resistance to ibrutinib, alternative non-covalent BTK inhibitors with activity against the C481S mutation are in development. In vitro studies of the BTK inhibitor ARQ531 demonstrate inhibition of BTK that harbors the C481S mutation [84]. Cytotoxicity with ARQ531 has been demonstrated in cells isolated from patients with the C481S mutations. In the TCL1 mouse model of CLL, ARQ531 demonstrated superiority to ibrutinib, in prolonging median survival of the mice. Likewise, REDX08608 is a next-generation BTK inhibitor that binds to C481S-mutated BTK that is currently being studied preclinically [85]. SNS-062 is a potent, non-covalent BTK inhibitor that has antitumor activity in vitro against tumor cells harboring the C481S mutation [86]. A phase Ia, first in human study in healthy subjects demonstrated reasonable safety with no grade 3 or higher toxicities observed. Common toxicities included headache, nausea, and constipation.
Whole-exome sequencing has also been performed on patients with acquired resistance to idelalisib [87]. Eighty-eight mutations were identified to be associated with progressive disease. However, no recurrent mutations were identified in more than one patient, and no mutations were identified in the PI3K signaling pathway or any other related pathway, suggesting that there is no common mutation mechanism for idelalisib resistance. In vivo studies using the murine CLL clone TCL1-192 have attempted to study resistance by exposing the tumor to the murine-specific PI3Kδ inhibitor GS-649443 [88]. Whole-exome sequencing of resistant TCL1-192 cells identified 64 specific mutations. As was seen in the samples taken from patients treated with idelalisib, no recurrent mutations were identified. However, a subset of mutations were functionally grouped into integrin and extracellular matrix signaling. All resistant tumors showed upregulation of genes involved in the integrin receptor complex. RNAseq analysis and Western blotting identified an upregulation of IGF1R expression in the resistant samples. Targeting IGF1R with linsitinib and GS-649443 resulted in improved survival of mice with resistant TCL1-192. Taken together, these studies suggest that resistance to PI3K inhibition is not mediated by unique, recurrent mutations in the BCR pathway but by alterations in survival signaling with potential contribution from various mutations.
Next-generation Inhibitors
Given the significant efficacy seen with ibrutinib and idelalisib, a number of next-generation agents are being investigated. In an effort to decrease off-target effects of BTK inhibition, many of the new inhibitors show increased specificity for BTK.
Next-generation BTK Inhibitors
Acalabrutinib binds covalently to the same binding site as ibrutinib but has increased specificity for BTK relative to the other Tec kinases [89]. Acalabrutinib’s IC50 for BTK is 5.1 vs. 1.5 nM for ibrutinib, 93 nM for TEC vs. 7 nM for ibrutinib, and greater than 1000 nM for ITK, SRC, and EGFR, against each of which ibrutinib has <20 nM potency. A phase I/II trial investigated the efficacy of acalabrutinib in 61 patients with relapsed or refractory CLL [89]. The overall response rate was 95% and the remaining 5% of patients had stable disease. In the subset of patients with 17p deletion, the response rate was 100%. One expansion cohort of the phase 1 acalabrutinib trial investigated patients with CLL who were intolerant to ibrutinib [90]. Thirty-three patients were enrolled. At a median follow-up of 9.5 months, 73% of patients continued on treatment. The most common adverse events were grade 1 and included diarrhea (42%), headache (39%), cough (24%), increased weight (24%), and nausea (21%). Two pneumonias, one grade 3, and the other grade 4 were the only serious adverse events occurring in more than one patient. No major bleeding occurred in the six patients who had developed bleeding while on ibrutinib. Patients discontinued acalabrutinib for progression (15%), adverse events (6%), and physician decision (6%). The two adverse events that resulted in cessation of acalabrutinib were thought to be unrelated to acalabrutinib and were a grade 5 fungal infection and a grade 3 metastatic endometrial cancer. An ongoing phase III trial is comparing acalabrutinib to ibrutinib in patients with relapsed high-risk disease defined by the presence of a deletion in 17p or 11q. Acalabrutinib alone or in combination with obinutuzumab is being compared to obinutuzumab chlorambucil in a three-arm trial of upfront treatment for patients with CLL (NCT02475681).
ONO/GS-4059 is a selective and irreversible inhibitor of BTK with decreased inhibition of EGFR and ITK relative to ibrutinib [91, 92]. ONO-4059 was investigated in a phase I study of patients with relapsed or refractory mature B cell malignancies. Twenty-four of 25 patients with CLL responded for an overall response rate of 96%, with an estimated PFS of 874 days. No diarrhea or cardiac events were reported but one patient had significant bleeding. GS-4059 is also being investigated in a phase Ib trial in combination with idelalisib for patients with relapsed or refractory B cell malignancies [93]. Twenty patients have been enrolled, eight with CLL. Ninety-five percent of patients reported a treatment emergent AE. Grade 3 or higher adverse events included neutropenia in five patients (25%), diarrhea in one patient (5%), and back pain in one patient (5%). Of the seven evaluable patients from the dose escalation portion of the trial, three have had a greater than 50% decrease in lymphadenopathy.
CC-292 is a rationally designed, potent, selective inhibitor of BTK that covalently binds to cysteine 481 on BTK, blocking the ATP-binding pocket of the enzyme [94]. A phase I, dose escalation study was performed in patients with relapsed or refractory CLL, B-NHL, or Waldenstrom’s macroglobulinemia [95]. Over 90% BTK receptor occupancy was observed with twice daily dosing. Eighty-four patients with CLL were treated. Among the CLL patients treated at the four highest dose levels, 60% of patients had a more than 50% reduction in lymph node diameter. Treatment was well tolerated and the maximum tolerated dose was not defined. Dose-limiting toxicities were seen in two patients experiencing grade 4 thrombocytopenia, one patient with grade 3 drug related pneumonitis, and one patient with reversible, grade 3 mental status changes. Febrile neutropenia was seen in only 2% of patients. The median response duration was 11.0, 5.6, not reached, and 8.4 months for the 750-mg and 1000-mg once daily and 375-mg and 500-mg twice daily groups, respectively. The reason for the decreased durability of response relative to ibrutinib is likely related to highly variable pharmacokinetics and pharmacodynamics.
BGB-3111 is a potent, highly specific, irreversible BTK inhibitor with increased selectivity relative to ibrutinib for BTK as compared to other members of the Tec kinase family, including ITK [96]. A phase I trial in patients with relapsed B cell malignancies, including 45 with CLL or SLL demonstrated median BTK occupancy of 99.5% with twice daily dosing. The response rate is 90%, with an additional 7% of patients having stable disease. No patients developed progression or Richter’s transformation. Three serious adverse events were reported, including one patient with grade 2 cardiac failure, one episode of grade 2 pleural effusion, and one case of grade 3 purpura (which was the only major bleeding event).
Next-generation PI3K Inhibitors
Pilaralisib (previously SAR245408) is a highly selective, reversible, and potent inhibitor of class I PI3k α, β, γ, and δ isoforms. In a phase I trial in patients with relapsed or refractory CLL and NHL [97], ten patients with CLL and 15 patients with NHL were treated. Five patients with CLL (50%) and three patients with NHL (20%) had a partial response. The most frequent adverse events were diarrhea (92%), pyrexia (52%), and fatigue (44%). Twenty percent of patients had grade 3 or higher diarrhea.
Copanlisib is another pan PI3K inhibitor with increased inhibition of α and δ subunits [98]. A phase II study investigated the efficacy of copanlisib in patients with CLL and NHL. Thirty-three patients were enrolled, including 13 with CLL. Across all disease groups, the overall response rate was 47%. The overall response rate for CLL was 38%, all of which were partial responses. The most common adverse events were hyperglycemia (70%), hypertension (70%) of which 49% were grade 3, fatigue (64%), diarrhea (36%), neutropenia (36%), and anemia (33%).
Duvelisib (previously IPI-145) is a dual PI3K p110 δγ inhibitor that was investigated in a phase I dose escalation trial in 54 patients [99]. Eighty-three percent of patients achieved a greater than 50% decrease in adenopathy by imaging, and best overall response rate by IWCLL criteria was 55% in 49 evaluable patients and included one complete response. The most common adverse events, of grade 3 or greater, included neutropenia (31%), febrile neutropenia (15%), and pneumonia (11%). Duvelisib has also been investigated in the upfront setting in combination with FCR for younger patients with CLL [100]. The initial analysis of a phase I trial evaluating this combination enrolled 12 patients with an overall response rate of 100% and a complete remission rate of 33%. Interestingly, the rate of MRD negativity in the bone marrow was 89% in the nine patients evaluated. The combination of duvelisib, rituximab, and bendamustine was investigated in 48 patients with relapsed NHL [101]. Eighteen CLL patients were treated in the dose expansion arm with an overall response rate of 92%.
TGR-1202 is a next-generation, once daily, oral PI3Kδ inhibitor. A phase 1 study in patients with CLL and NHL demonstrated reasonable safety and significant efficacy [102]. Toxicities included nausea (44%, grade 3/4 0%), diarrhea (36%; grade 3 or higher 1%), and fatigue (31%; grade 3 or higher 3%). Of note, the incidence of hepatotoxicity and colitis appeared to be significantly less than that reported with idelalisib. TGR-1202 has been used in combination with the CD20 antibody obinutuzumab and chlorambucil as treatment for patients with CLL [103]. Safety and efficacy were reported on 18 patients. The most common adverse events of grade 3 or 4 were neutropenia (61%), thrombocytopenia (33%), and elevations in transaminases (28%). Thirteen of 14 treatment naïve patients responded, four of which were complete responses. Two of three previously treated patients achieved a response. TGR-1202 has also been combined with ibrutinib as outlined above.
Conclusion
Inhibition of the BCR signaling pathway is now a standard part of the CLL armamentarium for the management of both upfront disease as well as the relapsed and refractory setting. Inhibitors of both BTK and PI3K have significant efficacy as single agents and when combined with chemo-immunotherapy. Clear differences in the toxicity profile between the two approved agents suggest that ibrutinib is the favored agent for the majority of patients. However, given the increased risk of bleeding and incidence of atrial fibrillation with ibrutinib, patients on blood thinners or at risk for atrial fibrillation may be initially treated with idelalisib as long as they are previously treated. Recent clinical trial data have demonstrated increased risk of death secondary to infections when idelalisib is used frontline. In addition, idelalisib has been shown to promote the development of immune-mediated hepatitis, colitis, and pneumonitis. With the understanding of the potential for these toxicities, it is important to monitor patients carefully, provide infectious prophylaxis, follow regular CMV PCRs, and manage neutropenia aggressively with growth factor support. The management and prevention of the autoimmune toxicities are summarized in a set of clinical guidelines [104].
Emerging understanding of the off-target effects of both BTK and PI3Kδ inhibition suggests the potential for more effective combinatorial strategies. In particular, compelling evidence suggests that BTK inhibition may have a role as an immune modulator that enhances the efficacy of immune therapy such as CAR T cells. While the current research environment surrounding idelalisib is complicated by the recent observation of increased infectious risk and immune-mediated toxicity, there remains the potential that these effects can be re-directed to assist immune-mediated treatments or even allow PI3K delta inhibitors to be used primarily as immunomodulatory therapies in solid tumors.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Dameshek W. Chronic lymphocytic leukemia—an accumulative disease of immunolgically incompetent lymphocytes. Blood. 1967;29(4):Suppl:566–84.
Rai KR. A critical analysis of staging in CLL. In: Chronic lymphocytic leukemia: recent progress and future direction. UCLA Symp Mol Cell Biol New Ser. 1987;59:253.
Fischer K, Cramer P, Busch R, Stilgenbauer S, Bahlo J, Schweighofer CD, et al. Bendamustine combined with rituximab in patients with relapsed and/or refractory chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2011;29(26):3559–66.
Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–74.
Tam CS, O'Brien S, Plunkett W, Wierda W, Ferrajoli A, Wang X, et al. Long-term results of first salvage treatment in CLL patients treated initially with FCR (fludarabine, cyclophosphamide, rituximab). Blood. 2014;124(20):3059–64.
Eichhorst B, Fink A-M, Busch R, Lange E, Köppler H, Kiehl M, et al. Chemoimmunotherapy with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) versus bendamustine and rituximab (BR) In: Previously untreated and physically fit patients (pts) with advanced chronic lymphocytic leukemia (CLL): results of a planned analysis. [Abstract 526] Blood. 2013;122(21).
Eichhorst B, Fink AM, Bahlo J, Busch R, Kovacs G, Maurer C, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016;17(7):928–42.
Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6.
Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–7.
Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208–15.
• Thompson PA, Tam CS, O'Brien SM, Wierda WG, Stingo F, Plunkett W, et al. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood. 2016;127(3):303–9. Demonstrated the potential for long term remission with FCR in patients with mutated IGVH.
Yamamoto T, Yamanashi Y, Toyoshima K. Association of Src-family kinase Lyn with B-cell antigen receptor. Immunol Rev. 1993;132:187–206.
Rolli V, Gallwitz M, Wossning T, Flemming A, Schamel WW, Zurn C, et al. Amplification of B cell antigen receptor signaling by a Syk/ITAM positive feedback loop. Mol Cell. 2002;10(5):1057–69.
Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116(12):2078–88.
Buchner M, Fuchs S, Prinz G, Pfeifer D, Bartholome K, Burger M, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69(13):5424–32.
Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309–12.
Chiorazzi N, Efremov DG. Chronic lymphocytic leukemia: a tale of one or two signals? Cell Res. 2013;23(2):182–5.
Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92.
Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117(23):6287–96.
Rozovski U, Harris DM, Li P, Liu Z, Jain P, Ferrajoli A, et al. Ibrutinib disrupts the metabolic program of CLL cells in-vivo. Blood. 2016;128(22):4348.
Herman SEM, Liu D, Landau DA, Sun C, Farooqui M, Wu CJ, et al. Dynamic alterations in gene expression in ibrutinib treated CLL reveal profound impact on multiple signaling pathways. Blood. 2016;128(22):189.
• Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2012;31(1):88–94. Ibrutinib activity in non-Hodgkin lymphoma.
• Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. Ibruitinib efficacy in CLL.
• Byrd JC, Brown JR, O'Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–23. Ibrutinib efficacy relative to ofatumumab in CLL.
• Brown JR, Hillmen P, O'Brien S, Barrientos JC, Reddy NM, Coutre SE, Tam CS, Mulligan SP, Jaeger U, Barr PM, Furman RR, Kipps TJ, Cymbalista F, Thornton P, Caligaris-Cappio F, Delgado J, Montillo M, DeVos S, Moreno C, Pagel JM, Munir T, Burger JA, Chung D, Lin J, Gau L, Chang B, Cole G, Hsu E, James DF, Byrd JC. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATETM study in patients with previously treated CLL/SLL. Leukemia. 2017. doi:10.1038/leu.2017.175.
Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16(2):169–76.
O'Brien S, Jones JA, Coutre SE, Mato AR, Hillmen P, Tam C, et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. Lancet Oncol. 2016;17(10):1409–18.
O'Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48–58.
Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–37.
Barr P, Robak T, Owen CJ, Tedeschi A, Bairey O, Bartlett NL, et al. Updated efficacy and safety from the phase 3 resonate-2 study: ibrutinib as first-line treatment option in patients 65 years and older with chronic lymphocytic leukemia/small lymphocytic leukemia. Blood. 2016;128(22):234.
Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2010;117(2):591–4.
• Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390–7. Idelalisib effective in CLL.
• Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997–1007. Idelalisib and rituximab more effective than rituximab alone in CLL.
Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370(11):1008–18.
Brown JR, Barrientos JC, Barr PM, Flinn IW, Burger JA, Tran A, et al. The Bruton tyrosine kinase inhibitor ibrutinib with chemoimmunotherapy in patients with chronic lymphocytic leukemia. Blood. 2015;125(19):2915–22.
Chanan-Khan A, Cramer P, Demirkan F, Fraser G, Silva RS, Grosicki S, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol. 2016;17(2):200–11.
Kipps TJ, Hillmen P, Demirkan F, Grosicki S, Coutre SE, Barrientos JC, et al. 11q Deletion (del11q) is not a prognostic factor for adverse outcomes for patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) treated with ibrutinib: pooled data from 3 randomized phase 3 studies. Blood. 2016;128(22):2042.
Burger JA, Keating MJ, Wierda WG, Hartmann E, Hoellenriegel J, Rosin NY, et al. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2014;15(10):1090–9.
Davids MS, Kim HT, Nicotra A, Savell A, Francoeur K, Hellman J, et al. TGR-1202 in combination with ibrutinib in patients with relapsed or refractory CLL or MCL: preliminary results of a multicenter phase I/Ib study. Blood. 2016;128(22):641.
Davids MS, Kim HT, Brander DM, Bsat J, Savell A, Francoeur K, et al. Initial results of a multicenter, phase II study of ibrutinib plus FCR (iFCR) as frontline therapy for younger CLL patients. Blood. 2016;128(22):3243.
Jones JA, Woyach J, Awan FT, Maddocks KJ, Whitlow T, Ruppert AS, et al. Phase 1b results of a phase 1b/2 study of obinutuzmab, ibrutinib, and venetoclax in relapsed/refractory chronic lymphocytic leukemia (CLL). Blood. 2016;128(22):639.
Zelenetz AD, Barrientos JC, Brown JR, Coiffier B, Delgado J, Egyed M, et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017;18(3):297–311
Zelenetz AD, Brown JR, Delgado J, Eradat H, Ghia P, Jacob A, et al. Updated analysis of overall survival in randomized phase III study of idelalisib in combination with bendamustine and rituximab in patients with relapsed/refractory CLL. Blood. 2016;128(22):231.
Fiorcari S, Brown WS, McIntyre BW, Estrov Z, Maffei R, O'Brien S, et al. The PI3-kinase delta inhibitor idelalisib (GS-1101) targets integrin-mediated adhesion of chronic lymphocytic leukemia (CLL) cell to endothelial and marrow stromal cells. PLoS One. 2013;8(12):e83830.
Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117(2):563–74.
Cheson BD, Byrd JC, Rai KR, Kay NE, O'Brien SM, Flinn IW, et al. Novel targeted agents and the need to refine clinical end points in chronic lymphocytic leukemia. J Clin Oncol. 2012;30(23):2820–2.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 Guidelines. Blood. 2008;111(12):5446–56.
Woyach JA, Smucker K, Smith LL, Lozanski A, Zhong Y, Ruppert AS, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2014;123(12):1810–7.
Quek LS, Bolen J, Watson SP. A role for Bruton's tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8(20):1137–40.
Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood. 2003;102(10):3592–9.
Hamazaki Y, Kojima H, Mano H, Nagata Y, Todokoro K, Abe T, et al. Tec is involved in G protein-coupled receptor- and integrin-mediated signalings in human blood platelets. Oncogene. 1998;16(21):2773–9.
Kunk PR, Mock J, Devitt ME, Palkimas S, Sen J, Portell CA, et al. Major bleeding with ibrutinib: more than expected. Blood. 2016;128(22):3229.
Pavlik A, Barr H, Dotson E, Byrd JC, Blum KA, Awan FT, et al. Major bleeding complications among patients treated with ibrutinib and concomitant antiplatelet, anticoagulant, or supplemental therapy. Blood. 2016;128(22):4387.
Kamel S, Horton L, Ysebaert L, Levade M, Burbury K, Tan S, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015;29(4):783–7.
Leong DP, Caron F, Hillis C, Duan A, Healey JS, Fraser G, et al. The risk of atrial fibrillation with ibrutinib use: a systematic review and meta-analysis. Blood. 2016;128(1):138–40.
Thompson PA, Levy V, Tam CS, al Nawakil C, Goudot FX, Quinquenel A, et al. The impact of atrial fibrillation on subsequent survival of patients receiving ibrutinib as treatment of chronic lymphocytic leukemia (CLL): An international study. Blood. 2016;128(22):3242.
Coutre S, Byrd JC, Hillmen P, Barrientos JC, Barr PM, Devereux S, et al. Integrated and long-term safety analysis of ibrutinib in patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL). Blood. 2016;128(22):4383.
Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539–49.
Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127(9):1117–27.
Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, Zhang Q, et al. The addition of the BTK inhibitor Ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin Cancer Res. 2016;22(11):2684–96.
Geyer MB, Park JH, Riviere I, Senechal B, Wang X, Purdon TJ, et al. Implications of concurrent ibrutinib therapy on CAR T-cell manufacturing and phenotype and on clinical outcomes following CD19-targeted CAR T-cell administration in adults with relapsed/refractory CLL. Blood. 2016;128(22):58.
Long M, Beckwith KA, Do P, Bethany ML, Gordon G, Lehman AM, et al. Ibrutinib represents a novel class of immune modulating therapeutics that enhances the survival of activated T cells in vitro and in vivo through a non-BTK mediated mechanism. Blood. 2016;128(22):3238.
Damle R, Schaffer M, Chaturvedi S, Phelps C, Aquino R, Mahler M, et al. Early changes in circulating T-cell immune profiles in patients with relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma: data from the phase 3, double-blind HELIOS trial. Blood. 2016;128(22):4397.
Biancotto A, Chang BY, Buggy JJ, McCoy JP, Farooqui M, Wiestner A. Cytokine and T-cell phenotypic changes upon in vivo ibrutinib therapy for CLL—targeting both CLL cells and the tumor-microenvironment. Blood. 2013;122(21):2856.
Kohrt HE, Sagiv-Barfi I, Rafiq S, Herman SE, Butchar JP, Cheney C, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123(12):1957–60.
Da Roit F, Engelberts PJ, Taylor RP, Breij EC, Gritti G, Rambaldi A, et al. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: implications for combination therapy. Haematologica. 2015;100(1):77–86.
Andrick B, Alwhaibi A, DeRemer D, Quershi S, Khan R, Shenoy S, et al. Antibody response to pneumococcal conjugate vaccine (PCV13) in chronic lymphocytic leukemia patients receiving ibrutinib. Blood. 2016;128(22):5597.
Rogers KA, Lehman AM, Cheney C, Goettl VM, Mantel R, Smith LL, et al. Inhibitors of Bruton's tyrosine kinase reduce anti-red blood cell response in a murine model of autoimmune hemolytic anemia. Blood. 2016;128(22):1259.
Manda S, Dunbar N, Marx-Wood CR, Danilov AV. Ibrutinib is an effective treatment of autoimmune haemolytic anaemia in chronic lymphocytic leukaemia. Br J Haematol. 2015;170(5):734–6.
St Bernard R, Hsia CC. Safe utilization of ibrutinib with or without steroids in chronic lymphocytic leukemia patients with autoimmune hemolytic anemia. Ann Hematol. 2015;94(12):2077–9.
Molica S, Levato L, Mirabelli R. Chronic lymphocytic leukemia, autoimmune hemolytic anemia and ibrutinib: a case report and review of the literature. Leuk Lymphoma. 2016;57(3):735–7.
Cavazzini F, Lista E, Quaglia FM, Formigaro L, Cavallari M, Martinelli S, et al. Response to ibrutinib of refractory life-threatening autoimmune hemolytic anemia occurring in a relapsed chronic lymphocytic leukemia patient with 17p deletion. Leuk Lymphoma. 2016;57(11):2685–8.
Lampson BL, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids MS, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood. 2016;128(2):195–203.
Yeung CCS, Hockenbery DM, Westerhoff M, Coutre S, Sedlak RH, Dubowy RL, et al. Pathology results of tissue biopsy during idelalisib-associated diarrhea/colitis. Blood. 2016;128(22):4391.
Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, et al. Cutting edge: the phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2006;177(10):6598–602.
Oak JS, Deane JA, Kharas MG, Luo J, Lane TE, Cantley LC, et al. Sjogren’s syndrome-like disease in mice with T cells lacking class 1A phosphoinositide-3-kinase. Proc Natl Acad Sci U S A. 2006;103(45):16882–7.
Yates J, Rovis F, Mitchell P, Afzali B, Tsang JY, Garin M, et al. The maintenance of human CD4+ CD25+ regulatory T cell function: IL-2, IL-4, IL-7 and IL-15 preserve optimal suppressive potency in vitro. Int Immunol. 2007;19(6):785–99.
Liu D, Zhang T, Marshall AJ, Okkenhaug K, Vanhaesebroeck B, Uzonna JE. The p110delta isoform of phosphatidylinositol 3-kinase controls susceptibility to Leishmania major by regulating expansion and tissue homing of regulatory T cells. J Immunol. 2009;183(3):1921–33.
Patton DT, Wilson MD, Rowan WC, Soond DR, Okkenhaug K. The PI3K p110delta regulates expression of CD38 on regulatory T cells. PLoS One. 6(3):e17359.
Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–94.
Liu TM, Woyach JA, Zhong Y, Lozanski A, Lozanski G, Dong S, et al. Hypermorphic mutation of phospholipase C, gamma2 acquired in ibrutinib-resistant CLL confers BTK independency upon B-cell receptor activation. Blood. 2015;126(1):61–8.
Woyach JA, Guinn D, Ruppert AS, Blachly JS, Lozanski A, Heerema NA, et al. The development and expansion of resistant subclones precedes relapse during ibrutinib therapy in patients with CLL. Blood. 2016;128(22):55.
Farag SM, Newton D, Doody G, Mahmoud LA, Fouda MI, El Ghannam DM, et al. Molecular mechanisms of ibrutinib resistance: defining a logical approach to improving targeted therapy in CLL. Blood. 2016;128(22):2046.
Reiff SD, Mantel R, Smith LL, McWhorter S, Goettl VM, Johnson AJ, et al. The Bruton's tyrosine kinase (BTK) inhibitor ARQ 531 effectively inhibits wild type and C481S mutant BTK and is superior to ibrutinib in a mouse model of chronic lymphocytic leukemia. Blood. 2016;128(22):3232.
Guisot NES, Best SA, Wright V, Thomason A, Woyach JA, Mantel R, et al. REDX08608, a Novel, potent and selective, reversible BTK inhibitor with efficacy and equivalent potency against wild-type and mutant C481S BTK. Blood. 2016;128(22):4399.
Neuman LL, Ward R, Arnold D, Combs DL, Gruver D, Hill W, et al. First-in-human phase 1a study of the safety, pharmacokinetics, and pharmacodynamics of the noncovalent Bruton tyrosine kinase (BTK) inhibitor SNS-062 in healthy subjects. Blood. 2016;128(22):2032.
Ghia P, Ljungström V, Tausch E, Agathangelidis A, Scheffold A, Scarfo L, et al. Whole-exome sequencing revealed no recurrent mutations within the PI3K pathway in relapsed chronic lymphocytic leukemia patients progressing under idelalisib treatment. Blood. 2016;128(22):2770.
Scheffold A, Jebaraj BMC, Tausch E, Yahiaoui A, Dolnik A, Blaette TJ, et al. In vivo modeling of resistance to PI3Kδ inhibitor treatment using EμTCL1-Tg tumor transfer model. Blood. 2016;128(22):190.
Byrd JC, Harrington B, O'Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323–32.
Awan FT, Schuh A, Brown JR, Furman RR, Pagel JM, Hillmen P, et al. Acalabrutinib monotherapy in patients with ibrutinib intolerance: results from the phase 1/2 ACE-CL-001 clinical study. Blood. 2016;128(22):638.
Walter HS, Rule SA, Dyer MJ, Karlin L, Jones C, Cazin B, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood. 2016;127(4):411–9.
Liclican A, Xing W, Serafini L, Wang T, Brendza K, Lutz J, et al. Biochemical characterization of GS-4059 as a potent and selective covalent irreversible inhibitor of Bruton’s tyrosine kinase. Blood. 2016;128(22):1594.
Salles GA, Morschhauser F, Cheson B, Rule SA, Fegan C, Guillaume C, et al. Preliminary results of a phase 1b dose escalation and dose expansion study of GS-4059 in combination with idelalisib in subjects with B-cell malignancies. Blood. 2016;128(22):2961.
Arnason JE, Brown JR. B cell receptor pathway in chronic lymphocytic leukemia: specific role of CC-292. Immunotargets Ther. 2014;3:29–38.
Brown JR, Harb WA, Hill BT, Gabrilove J, Sharman JP, Schreeder MT, et al. Phase I study of single-agent CC-292, a highly selective Bruton’s tyrosine kinase inhibitor, in relapsed/refractory chronic lymphocytic leukemia. Haematologica. 2016;101(7):e295–8.
Tam CS, Opat S, Cull G, Trotman J, Gottlieb D, Simpson D, et al. Twice daily dosing with the highly specific BTK inhibitor, Bgb-3111, achieves complete and continuous BTK occupancy in lymph nodes, and is associated with durable responses in patients (pts) with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL). Blood. 2016;128(22):642.
Brown JR, Davids MS, Rodon J, Abrisqueta P, Kasar SN, Lager J, et al. Phase I trial of the pan-PI3K inhibitor pilaralisib (SAR245408/XL147) in patients with chronic lymphocytic leukemia (CLL) or relapsed/refractory lymphoma. Clin Cancer Res. 2015;21(14):3160–9.
Dreyling M, Cunningham D, Bouabdallah K, Assouline S, Van den Neste E, Vitolo U, et al. Phase 2A Study of Copanlisib, a Novel PI3K Inhibitor, in Patients with Indolent Lymphoma. Blood. 2014;124(21):1701.
O'Brien S, Patel M, Kahl BS, Horwitz SM, Foss FM, Porcu P, et al. Duvelisib (IPI-145), a PI3K-δ,γ inhibitor, is clinically active in patients with relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;124(21):3334.
Davids MS, Kim HT, Gilbert E, Cowen L, Francoeur K, Hellman J, et al. Preliminary results of a phase Ib study of duvelisib in combination with FCR (dFCR) in previously untreated, younger patients with CLL. Blood. 2015;126(23):4158.
Flinn IW, Cherry M, Maris M, Matous JV, Berdeja JG, Patel MR. Combination trial of duvelisib (IPI-145) with bendamustine, rituximab, or bendamustine/rituximab in patients with lymphoma or chronic lymphocytic leukemia. Blood. 2015;126(23):3928.
O'Connor OA, Flinn IW, Patel MR, Fenske TS, Deng C, Brander DM, et al. TGR-1202, a novel once daily PI3K-delta inhibitor, demonstrates clinical activity with a favorable safety profile in patients with CLL and B-cell lymphoma. Blood. 2015;126(23):4154.
Mahadevan D, Pauli EK, Cutter K, Dietz LA, Sportelli P, Miskin HP, et al. A phase I trial of TGR-1202, a next generation once daily PI3K-delta inhibitor in combination with obinutuzumab plus chlorambucil, in patients with chronic lymphocytic leukemia. Blood. 2015;126(23):2942.
Coutre SE, Barrientos JC, Brown JR, de Vos S, Furman RR, Keating MJ, et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma. 2015;56(10):2779–86.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Jon E. Arnason has received compensation from Gilead for service on an advisory board.
Jennifer R. Brown has received compensation from Janssen, Gilead, Celgene, Sun BioPharma, Novartis, AbbVie, Pfizer, AstraZeneca, Astellas, Redx, Pharmacyclics, and Genentech/Roche for service as a consultant.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Lymphomas
Rights and permissions
About this article

Cite this article
Arnason, J.E., Brown, J.R. Targeting B Cell Signaling in Chronic Lymphocytic Leukemia. Curr Oncol Rep 19, 61 (2017). https://doi.org/10.1007/s11912-017-0620-7
Published:
DOI: https://doi.org/10.1007/s11912-017-0620-7