
Abstract
Recurrent ligand-binding domain ESR1 mutations have recently been detected in a substantial number of patients with metastatic ER+ breast cancer and evolve under the selective pressure of endocrine treatments. In this review, we evaluate the current understanding of the biological and clinical significance of these mutations. The preclinical studies revealed that these mutations lead to constitutive ligand-independent activity, indicating resistance to aromatase inhibitors and decreased sensitivity to tamoxifen and fulvestrant. Retrospective analyses of ESR1 mutations in baseline plasma circulating tumor DNA from completed clinical trials suggest that these mutations are prognostic and predictive of resistance to aromatase inhibitors in metastatic disease. Currently, we are lacking prospective studies to confirm these results and to determine the optimal treatment combinations for patients with the ESR1 mutations. In addition, the clinical development of novel agents to overcome resistance engendered by these mutations is also needed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple epidemiological, molecular, and clinical studies suggest a key role for the female hormone estradiol and its cognate receptor, estrogen receptor α (ER), in the development and progression of breast cancer. ER-positive (ER+) breast cancers, which constitute about 70% of all breast cancers, express the ER protein. ER+ breast cancers are classified molecularly as mainly the luminal subtype, which includes the more indolent and endocrine-sensitive luminal A and the more aggressive and less endocrine-sensitive luminal B subtypes [1]. ER is a ligand-dependent transcription factor and a member of the nuclear receptor superfamily. The protein consists of multi-functional domains including the DNA-binding and hinge domains flanked by the transcriptional transactivation domains, the ligand-independent activating function-1 (AF-1) and the ligand-dependent activating function-2 (AF-2). The AF2 overlaps with the ligand-binding domain (LBD). Upon ligand binding, ER undergoes a conformational change, dimerizes, and is recruited to numerous DNA sites to regulate the transcription of genes involved in physiological and cancer processes. The genome-wide distribution of ER-binding sites within a particular cell (termed the ER cistrome) and its transcriptional activity are influenced by receptor tyrosine kinase pathways, other signaling networks, the repertoire of the ER-co-regulatory (co-activators and co-repressors) and pioneer factors, and chromatin accessibility [2, 3].
Endocrine Therapy and Mechanisms of Endocrine Resistance
The most commonly used endocrine (hormonal) therapies inhibit ER activity by either targeting the ER protein itself or depriving the receptor of its ligand. The different classes of endocrine treatments include selective ER modulators (SERMs; e.g., tamoxifen), which compete with the estrogen ligand and have mixed agonistic/antagonistic capacities, selective ER degraders (SERDs; e.g., fulvestrant), which possess almost exclusive antagonistic activity and induce ER protein degradation, and treatment strategies that deplete systemic estrogen levels by either aromatase inhibitors or pharmacological or surgical ovarian ablation in post- and premenopausal women, respectively. While endocrine therapies have proven to be very effective in both the early and the metastatic settings, both de novo and acquired resistance to endocrine treatments remain a key clinical challenge [4].
Preclinical and clinical studies have elucidated a number of potential mechanisms underlying endocrine resistance. These mechanisms involve pathways that either interact with ER, its co-regulators, or other transcriptional factors to alter ER activity and sensitivity to endocrine treatments or escape pathways that can bypass ER dependency by providing alternative, proliferative, and survival stimuli to sustain tumor growth and progression. Modulations of these pathways can be driven by genomic, epigenetic or tumor microenvironment influences. A detailed discussion on these mechanisms is beyond the scope of this review and can be found elsewhere [5, 6].
A complete loss of ER is uncommon and observed in clinical samples in 10–20% of metastatic breast cancers [7]. The fact that many tumors with acquired endocrine resistance can still respond to additional lines of endocrine therapies suggests that ER continues to play a key role, albeit an altered one in these tumors. Increased ligand-independent activity or decreased response to SERMs and SERDs can be facilitated by multiple posttranslational modifications of ER as a result of increased growth factor and other kinase signaling pathways. Changes in the ratio of the levels of positive and negative ER co-regulators or in their activity have also been shown to play a role in endocrine resistance [8, 9]. Amplification of the ESR1 gene that encodes the ER or ESR1 gene fusions may be key drivers of resistance in some tumors, although these genetic alterations are relatively uncommon [10, 11]. In contrast, recurrent activating missense mutations clustered within the ER LBD have been recently reported by multiple groups and found in a substantial fraction of endocrine-resistant metastatic ER+ breast cancers [10, 12, 13, 14, 15••, 16].
ESR1 Ligand-Binding Mutation Missense Mutations in Breast Cancer Tissue Samples
The ESR1 Y537N-activating missense mutation was first described in a single metastatic sample in 1997 [17]. But other studies, mainly in treatment-naïve primary tumors, failed to detect this or similar ESR1 mutations [18, 19]. Since 2013, a series of studies using next-generation sequencing reported the detection of ESR1 recurrent LBD mutations in metastatic ER+ breast cancer tissue specimens (Fig. 1a). The first study employed whole exome sequencing of patient-derived xenograft (PDX) models derived from ER+ metastatic samples. ESR1 mutations were found in three of the seven models and were corroborated with the originating metastatic tumors [10, 12, 13].

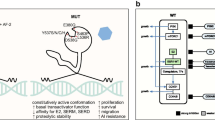
a Estrogen receptor α (ER) structural/functional domains and the distribution of the somatic ESR1 ligand-binding domain (LBD) point mutations identified in tissue specimens of ER-positive breast tumors by next-generation sequencing in recent reports (references 10, 12, 13, 14, 15••). Each mutation is represented by a red bar at the specific ER protein position. b Direct consequences of the conformational changes in the ER LBD mutations: estrogen (E2) binding to LBD of the wild-type (WT) ER leads to receptor dimerization and stabilization of the α-helix of helix 12 (H12) in an active conformation, thus allowing the binding of co-activators (CoA) [top panel]. The Y537S and D538G mutations, denoted by red X, lead to a conformational change in the H12 that promotes stabilization in the agonist mode. This conformational change enables CoA binding in the absence of ligand. The conformational changes also lead to a decreased affinity for tamoxifen and decreased potency of tamoxifen. The conformational changes of D538G are different from Y537S and result in more subtle stabilization of H12 and lower binding affinity of the ER co-activator AIB1 when compared to Y537S [bottom panel]. AF-1 activation function-1, AF-2 activation function-2, LBD ligand-binding domain
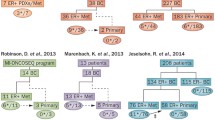
A second study included two cohorts of ER+ metastatic tumors [12]. One of the cohorts consisted of 44 patients from the BOLERO-2 study, which accrued patients that developed disease resistant to aromatase inhibitor (AI) treatment either within 12 months of completion of adjuvant AI treatment or disease progression on AI treatment in metastatic disease. The other cohort included 36 patients that developed disease progression after at least 3 months of endocrine therapy. Using targeted next-generation sequencing, this study found 14 patients with ESR1 LBD mutation among the 80 patients (17.5%). Additionally, sequencing of 183 primary tumors of a subset of the BOLERO-2 patients with metastatic disease detected mutations in six of these primary tumors (3.3%).
In addition, in two small studies in which 11 and 13 metastatic ER+ tumor samples were sequenced, ESR1 LBD recurrent mutations were found in six and five tumors, respectively [13, 14]. These two studies included together a subset of eight matched primary tumors in which no ESR1 LBD mutations were detected in the primary tumors. In another larger study, 76 metastatic ER+ metastatic tumors were sequenced and 11 ESR1 LBD mutations were detected (14.4%) [15••]. Finally, in a more recent study, where ESR1 mutations were assessed using droplet digital (dd) PCR in 37 ER+ matched metastatic and primary tumor samples, four ESR1 LBD mutations were found only within the metastatic tumors (10.8%) [16].
Similarly, in all the studies mentioned above, with the exception of the BOLERO-2 cohort, in the limited available matched primary tumors and in a small number of additional primary tumors without matched metastatic samples, the ESR1 mutations were not detected. In addition, in the provisional TCGA study and Metabric study available online on the cBioPortal [20, 21], which used next-generation sequencing, ESR1 mutations were detected in less than 1% of primary tumors. In contrast, two recent studies using the more sensitive dd-PCR approach to detect the recurrent LBD mutations with larger sample numbers of primary tumors found higher percentages of ESR1 mutations. In the first study, ESR1 mutations were detected in 2.5% of 270 primary tumors. This study also detected 11 ESR1 mutations in 55 (20%) ER+ metastatic tissue samples by dd-PCR [22]. In the second study, 203 primary tumors were analyzed by dd-PCR and a relatively high percentage of these tumors harbored LBD ESR1 mutations (the Y537N mutation was found in 12% of the tumors, the Y537S in 5%, and the D538G mutation in 2%) [23]. Of note, in this study, the LBD mutations in the primary tumors were not associated with long-term outcomes after adjuvant tamoxifen treatment [23].
The Biology of the ESR1 Ligand-Binding Domain Mutations
The recurrent ESR1 mutations detected in ER+ breast cancers were all found within the LBD. The most common mutations were mutations in the Y537 and D538 amino acid residues. Preclinical studies revealed that these are gain-of-function mutations that lead to constitutive, ligand-independent activation of ER (Fig. 1b) [12, 14, 15••]. In addition, these mutations lead to relative resistance to tamoxifen and fulvestrant. The Y537 and D538 mutations reside in helix 12 (H12), a key structural part of AF2. Estrogen binding to the LBD leads to stabilization of H12 in an active conformation, allowing the binding of co-activators, such as NCOA3, and results in activation of the receptor. In contrast, the binding of an antagonist to the LBD induces an H12 inactive conformation that promotes co-repressor binding and inhibition of the receptor activity.
Early structural studies have shown that the Y537S ER mutation stabilizes H12 in the agonistic conformation, similar to wild-type (WT)-ER when bound to E2 [24]. More recent simulation studies and a crystallography study of the D538G-mutant structure show that this mutation also stabilizes H12 in the agonistic conformation in the absence of E2, albeit less potently when compared to the Y537S mutation [25••]. In addition, the crystal structure and simulation studies of tamoxifen bound to the D538G and Y537S mutants indicate that these mutations confer an altered antagonistic conformation, facilitating resistance to antagonism. Affinity studies show that these mutated receptors have a decreased affinity for tamoxifen and E2. Finally, several studies showed increased co-activator binding to mutant ER when compared to WT-ER under ligand-independent conditions or in the presence of tamoxifen [12, 25••]. Together, these observations provide a mechanistic explanation for the ligand-independent activity and the reduced inhibitory potency of SERMs like tamoxifen.
The LBD ER mutants engender a growth advantage in estrogen-independent conditions and relative resistance to tamoxifen and fulvestrant. These phenotypes were shown in vitro as well as in vivo using breast cancer cell lines engineered to express the mutations and PDXs derived from human metastatic breast cancer tumors harboring the LBD mutations [10, 12, 15••]. In addition, an increased migratory capacity was observed in breast cancer cells with the D538G mutation, suggesting an enhanced invasive phenotype, which could explain the propensity of these mutations in the metastatic setting [14]. Gene expression and signaling analysis indicated that the mutations activate a unique set of genes and enhance the cross talk with IGF signaling [12, 23]. Whether and how these or other molecular changes mediate a metastatic potential remain elusive and are of importance. In addition, most of the mechanistic in vitro studies have focused on the most prevalent mutations. The E380Q mutation is another recurrent LBD ESR1 detected less frequently than the mutations in Y537 and D538. A PDX model with this mutation remained ligand dependent, and in one study, a non-breast cancer cell line expressing this mutation did not exhibit constitutive transcriptional activity [10, 26]. Thus, this mutation may facilitate tumor growth through a different mechanism and additional studies are needed to fully characterize the phenotypes engendered by this mutation.
ESR1 Mutations in Circulating Tumor DNA
Overall, the aforementioned studies of tumor tissue specimens showed that a significant percentage of patients with metastatic ER+ breast cancer harbor recurrent ESR1 LBD mutations. The high prevalence of these mutations and their biology imply that these mutations play an important role in resistance to treatment and disease progression. However, to further validate the clinical impact of these mutations and eventually use these mutations as biomarkers for treatment decision, a non-invasive assay to detect and serially monitor these mutations is needed. In addition, breast cancers are heterogeneous and in many cases a single biopsy cannot depict the full genomic landscape of the disease. Therefore, a number of groups have been developing non-invasive liquid biopsies to detect the ESR1 and other mutations in circulating tumor DNA (ctDNA) isolated from plasma samples of breast cancer patients, especially with metastatic disease.
Numerous recent studies have demonstrated the detection of mutant DNA alleles in ctDNA isolated from plasma as a tumor-specific biomarker [27, 28]. One of the seminal studies looked at serial levels of PIK3CA and TP53 mutations, which are prevalent mutations in primary and metastatic breast cancers [29]. In a cohort of 30 patients with metastatic breast cancer who were found to have a PIK3CA or TP53 through analysis of the archival primary tumor tissue specimens, ctDNA was detected in 97% of the patients. The single patient in which ctDNA was not detected had limited tumor burden and stable disease throughout the study. When compared to other circulating biomarkers, such as CA15-3 levels and circulating tumor cells, ctDNA was found to be more sensitive and had a greater dynamic range that correlates with changes in tumor burden. Moreover, increasing levels of ctDNA was prognostic in this study.
A number of groups investigated ctDNA ESR1 LBD mutations in order to develop a non- invasive reliable method for the detection of ESR1 mutations and to study in a more definitive manner the association between the presence of the ESR1 mutations and response to endocrine treatments and other targeted treatments. In one ctDNA study, ESR1 mutations were found in 25% of advanced breast cancer patients (9/29) and serial monitoring displayed changes in allele frequency (AF) [22]. In another study, plasma was tested for ESR1 LBD mutations (L536R, Y537C, Y537N, D538G) using dd-PCR in 128 patients with advanced ER+ breast cancer at the time of disease progression [30••]. ESR1 LBD mutations were found in 14.8% of these patients and were found exclusively in patients who had prior treatment with an AI. An analysis of a limited number of patients with and without detectable ctDNA ESR1 mutations showed that the ESR1 mutations are associated with a shorter progression-free survival (PFS) on subsequent AI treatment. Interestingly, patients who received an AI in the adjuvant setting or developed recurrent disease on adjuvant AI treatment had a lower prevalence of ESR1 mutations compared to patients that received AI treatment for metastatic disease only (5.8 and 4.8%, respectively, versus 36.4%). The authors hypothesized that this difference is due to the lack of genetic diversity in the presence of micrometastatic disease. These results have important clinical implications, but this study was relatively small, included a non-stratified cohort of patients with metastatic disease, and did not look at the associations between the ESR1 mutations and other endocrine treatments or other targeted therapies.
More recently, three groups performed post hoc prospective-retrospective analyses of ESR1 mutations in plasma samples from completed randomized clinical trials (Table 1) [26, 31••, 32]. In the first study, blood samples from the FERGI study were analyzed [26]. The FERGI study was a randomized phase II study that compared pictilisib, a pan-PI3K kinase inhibitor, plus fulvestrant with fulvestrant plus placebo in patients with metastatic ER+ breast cancer who progressed on AI treatment. Baseline ctDNA was analyzed for 12 ESR1 (E380Q, S463P, V534E, P535H, L536P, L536H, L536H, Y536S, Y536N, Y537C, D538G) and 9 PIK3CA mutations. ESR1 mutations were detected in 37% of plasma samples (78/207). In this study, ESR1 mutations were enriched in patients with luminal A tumors compared to luminal B and in PIK3CA-mutated tumors. Importantly, ESR1 mutations were not associated with differential PFS with the fulvestrant plus placebo or the fulvestrant plus pictilisib arm. In addition, serial testing of ctDNA was performed in 71 of the patients. Clinical response was associated with decreases in the AF of ESR1 and PIK3CA mutations, and increases in AF were seen only in patients with stable disease (SD) or progressive disease (PD). However, these changes did not reliably predict response, as there were patients with SD or PD in which a decrease in AF was measured.
In the second study, baseline ctDNA for seven ESR1 mutations (E380Q, L536R, Y537C, D538G, S463P, Y537N, Y537S) was evaluated in two phase III clinical trials in patients with metastatic ER+ breast cancer that progressed on endocrine treatment, including the SOFEA and PALOMA3 studies [31••]. In the SOFEA study, patients that had demonstrated prior sensitivity to an AI, in the adjuvant or metastatic setting, were randomized to fulvestrant plus anastrozole, fulvestrant plus placebo, or exemestane. ESR1 mutations were detected in 39.1% of a relatively small subgroup of patients in this trial (63/161). Patients with an ESR1 mutation had improved PFS with a fulvestrant-containing treatment (N = 45) compared to exemestane alone (N = 18), whereas patients with WT ESR1 had similar PFS on either treatment. However, the interaction test between ESR1 mutation and relative sensitivity to fulvestrant over exemestane was not statistically significant. In the PALOMA3 trial, patients who had prior endocrine treatment (tamoxifen or an AI) were randomized to receive fulvestrant plus palbociclib, a CDK4/6 inhibitor, or fulvestrant plus placebo. ctDNA for ESR1 mutations was tested in baseline plasma samples and found in 25.3% of the cohort that was tested (91/360). The PFS with the combination treatment (N = 63) was improved compared to fulvestrant plus placebo in patients with ESR1 mutations (N = 28) and patients with WT ESR1. In PALOMA3, overall, patients with ESR1 mutations had worse PFS compared to WT ESR1 patients. In addition, in this study, ESR1 mutations were almost exclusively found in patients with prior AI treatment, rare in patients that had prior tamoxifen only, and associated with prior response to endocrine treatment.
Lastly, a third study tested baseline ctDNA for ESR1 mutations (D538G, Y537S) in the BOLERO2 study [32]. This was a phase III study in which patients with metastatic ER+ breast cancer and prior exposure to a non-steroidal AI were randomized to exemestane versus exemestane plus the mTOR inhibitor everolimus. In this study, ESR1 mutations were detected in 28.8% of patients (156/541). Patients with an ESR1 mutation had worse overall survival (OS) compared to WT ESR1. A subgroup analysis showed that patients with the D538G mutation alone, the Y537S mutation alone, or both mutations all had decreased OS compared to WT ESR1. In contrast to the SOFEA study, in this study, the presence of the D538G mutation only (N = 24) was associated with decreased PFS in the single-agent exemestane treatment arm. As for the benefit from everolimus, the presence of the D538G mutation (N = 59) was not associated with differential response to everolimus plus exemestane, whereas the presence of a Y537S mutation (N = 21) or both mutations was associated with worse PFS. Similar to the previous studies, in the BOLERO2 study, the prevalence of the ESR1 mutations was higher in patients that received prior endocrine treatment in the metastatic setting (33%) compared to those that received endocrine treatment in the adjuvant setting only.
Taken together, in these studies, the prevalence of the ESR1 mutations determined by ctDNA in patients with metastatic ER+ breast cancer is in the range of 25–40%. The ESR1 mutations are associated with prior exposure to AI treatment. In addition, the presence of ctDNA ESR1 mutations is associated with poor outcomes. Given the retrospective and exploratory nature of these analyses and the relatively small number of patients with ESR1 mutations in the different treatment arms, it is hard to make strong conclusions regarding the associations between the ESR1 mutations and clinical response to treatment. Prospective studies in which patients are stratified based on ESR1 mutations are still needed to elucidate the impact of these mutations on response to different subsequent endocrine treatments and other targeted treatments.
Tumor Evolution and Clinical Implications
The need for cancer cells to resist and adapt to signals from the microenvironment and other exogenous factors, including treatment interventions, and the genetic instability in cancers resulting in the accumulation of somatic mutations is the basis for clonal evolution in cancers. Preclinical and clinical studies also suggest that acquired resistance to cancer treatments is due to the expansion of rare pre-existing (or acquired) drug-resistant clones under the selective pressure of targeted treatments [33, 34]. The ligand-independent and endocrine-resistant properties of the ESR1 LBD mutations and the fact that they are more prevalent in metastatic disease after treatment with aromatase inhibitors indicate that the selection of these mutations occurs under the pressure of endocrine treatment, particularly aromatase inhibitors. In addition, the wide range of allele frequencies and the polyclonality detected in tumor tissues and ctDNA imply that there is continued tumor heterogeneity even after the clonal selection.
The ESR1 LBD mutations were either undetectable or found at a very low frequency in primary tumors to date, suggesting that the mutations are either acquired or arise from a rare clone. In a more recent study that applied a more sensitive assay, ESR1 mutations were found in higher percentage of primary tumors [23], suggesting that with deeper sequencing methods, the ESR1 mutations might be present at a higher prevalence in primary tumors. The presence of ESR1 mutations in primary tumors may have an impact on the risk of disease recurrence and choice of adjuvant treatments, and therefore, it is important to determine the frequency in primary tumors. On the other hand, there is some evidence that AI treatment in the adjuvant setting does not strongly impact the evolution of the ESR1 mutations, hypothesized to be due to the low tumor burden in the micrometastatic setting [30••]. This raises the question of the significance of the presence of the ESR1 mutations in primary tumors and the implications in the decision-making of specific adjuvant endocrine treatment in the context of the ESR1 mutations. Resolution of this important issue awaits analysis of large adjuvant endocrine therapy trials.
In metastatic disease, multiple studies show that the prevalence of the ESR1 mutations is substantial and that the detection of these mutations is overall prognostic of poor outcome. In this setting, ctDNA ESR1 mutation testing is a sensitive and specific tool. However, the published studies to date on ESR1 mutations in ctDNA were conducted in individual research laboratories and differed in the specific mutations that were assayed, and a standardized test with a standardized cutoff has not been established. In addition, the majority of the studies used dichotomized results of baseline ctDNA analysis and it is not known if the AF and kinetics of the AF based on serial testing are of clinical significance. Another limitation of the recent post hoc studies is the retrospective design of these studies and lack of power, making the results hypothesis-driving only.
Given these limitations, it is not surprising that there are some discrepancies between the studies. As an example, in the analysis of the SOFEA study, patients with any ESR1 mutation had worse PFS with exemestane compared to patients with WT ESR1, whereas in the BOLERO2 study, only the patients with the D538G mutation had worse PFS on exemestane when compared to WT ESR1. Randomized studies stratified by patients with the ESR1 mutations are needed to determine if patients with the ESR1 mutations are less sensitive to exemestane and if the clinical benefit from fulvestrant at the current dose is similar compared to patients with WT ESR1. In addition, since everolimus, palbociclib, and more recently the CDK4/6 inhibitor ribociclib [35] are approved drugs in metastatic ER+ breast cancer, it is important to study the sensitivity of the mutations to these treatments and, not less important, the effect of these treatments on the evolution of the mutations. Another open question that is important to sort out is the sensitivity of the ESR1-mutant breast cancers to tamoxifen, since tamoxifen might be a better option than aromatase inhibitors in the presence of the mutations, particularly in combination with the other targeted agents.
Novel therapeutic strategies based on ongoing preclinical studies should also be considered in the future. The preclinical studies show that the mutant ER has lower affinity to tamoxifen and fulvestrant, suggesting that high doses might be more effective. Alternatively, newer SERMs/SERDs, such as bazedoxifene [36], RAD1901 [37], or GDC-0810 [38], might have a better binding affinity for the mutant ER and are currently under investigation. Bazedoxifene is a third-generation SERM with SERD activity. In a preclinical study of a PDX model harboring the Y537S, bazedoxifene as a single agent or in combination with palbociclib effectively reduced tumor growth [39]. We are currently conducting a phase Ib/II clinical trial with the combination of bazedoxifene and palbociclib for patients with metastatic breast cancer (NCT02448771). This trial includes serial monitoring of ctDNA ESR1 mutations.
GDC-0810 is a new orally available SERD that is currently being investigated in combination with palbociclib (NCT01823835). In preclinical studies, GDC-0810 had decreased potency in a cell line model of the Y537S but was effective in inhibiting the growth of an Y537S ER-mutant PDX [40]. Since the mutant ER activity remains dependent on co-activator binding, including NCOA3, drugs that target co-activators might offer another therapeutic option [25••, 41]. Lastly, similar to some other missense mutations, the ESR1 LBD mutations may be immunoreactive and could be leveraged by personalized vaccinations or candidates for treatments with immune modulators, potentially offering a wide range of therapeutics.
Conclusions
The recent discovery that LBD-activating ESR1 mutations are present in a high percentage of metastatic ER+ breast cancers sheds new light on the mechanisms of endocrine resistance. Based on the analysis of tissue specimens, the mutations are found in between 15 and 20% of metastatic ER+ tumors, with a relatively higher prevalence in patients who have received multiple lines of endocrine treatment. The analysis of ctDNA, which can capture the complexity of intra-tumoral heterogeneity and the heterogeneity between different metastatic sites, has found that these ESR1 mutations are detectable in up to 40% of patients with metastatic ER+ breast cancer. Testing ctDNA for ESR1 mutations in the metastatic setting is a promising tool for the continued investigation of the clinical role of these mutations as a predictive biomarker; however, ultimately for clinical utility, this test will need to be standardized.
The preclinical studies show that these mutations lead to ligand-independent constitutive activity and reduced sensitivity to tamoxifen and fulvestrant. In line with the preclinical data, the recent post hoc analyses of ESR1-mutant ctDNA in randomized clinical trials show that in patients with metastatic breast cancer, after first line treatment with a non-steroidal AI, patients with an ESR1 mutation have worse PFS on a steroidal AI compared to patients with WT ESR1. In addition, ESR1-mutant metastatic breast cancer patients seem to benefit from fulvestrant as a single agent or in combination with palbociclib. The dose of fulvestrant in these clinical trials may be sufficient to overcome at least the relative resistance predicted by the preclinical studies.
These retrospective analyses are exploratory only and are not practice changing. Prospective clinical trials in which patients are stratified on the basis of the ESR1 mutant status are needed, both to confirm these findings and to investigate the sensitivity of the mutations to other drugs such as tamoxifen, everolimus, and ribociclib. In addition, clinical trials dedicated to patients with ESR1 mutations will be needed in order to study novel agents to overcome the resistance engendered by these mutations that are currently under investigation in preclinical models.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74.
Carroll JS, Meyer CA, Song J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–97.
Lupien M, Meyer CA, Bailey ST, et al. Growth factor stimulation induces a distinct ER(alpha) cistrome underlying breast cancer endocrine resistance. Genes Dev. 2010;24:2219–27.
Williams N, Harris LN. The renaissance of endocrine therapy in breast cancer. Curr Opin Obstet Gynecol. 2014;26:41–7.
Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–43.
Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–47.
Hoefnagel LD, Moelans CB, Meijer SL, et al. Prognostic value of estrogen receptor alpha and progesterone receptor conversion in distant breast cancer metastases. Cancer. 2012;118:4929–35.
Torres-Arzayus MI, Font de Mora J, Yuan J, et al. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;6:263–74.
Schiff R, Massarweh S, Shou J, Osborne CK. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin Cancer Res. 2003;9:447S–54S.
Li S, Shen D, Shao J, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4:1116–30.
Veeraraghavan J, Tan Y, Cao XX, et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun. 2014;5:4577.
Toy W, Shen Y, Won H, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–45.
Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51.
Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, et al. D538G mutation in estrogen receptor-alpha: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013;73:6856–64.
•• Jeselsohn R, Yelensky R, Buchwalter G, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–67. ESR1 LBD mutations are detected in metastatic ER+ breast cancers only and the prevalnce is associated with number of treatment lines in the metastatic setting.
Fumagalli D, Wilson TR, Salgado R, et al. Somatic mutation, copy number and transcriptomic profiles of primary and matched metastatic estrogen receptor-positive breast cancers. Ann Oncol. 2016;27:1860–6.
Zhang QX, Borg A, Wolf DM, et al. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997;57:1244–9.
Karnik PS, Kulkarni S, Liu XP, et al. Estrogen receptor mutations in tamoxifen-resistant breast cancer. Cancer Res. 1994;54:349–53.
Roodi N, Bailey LR, Kao WY, et al. Estrogen receptor gene analysis in estrogen receptor-positive and receptor-negative primary breast cancer. J Natl Cancer Inst. 1995;87:446–51.
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Wang P, Bahreini A, Gyanchandani R, et al. Sensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patients. Clin Cancer Res. 2016;22:1130–7.
Gelsomino L, Gu G, Rechoum Y, et al. ESR1 mutations affect anti-proliferative responses to tamoxifen through enhanced cross-talk with IGF signaling. Breast Cancer Res Treat. 2016;157:253–65.
Nettles KW, Bruning JB, Gil G, et al. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat Chem Biol. 2008;4:241–7.
•• Fanning SW, Mayne CG, Dharmarajan V et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 2016; 5. The ESR1 ligand-binding mutations Y537S and D538G have decreased affinity to estradiol and tamoxifen and ligand-independent binding to AIB1 which provides a mechanistic explanation to the phenotypes of these mutations.
Spoerke JM, Gendreau S, Walter K, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun. 2016;7:11579.
Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224.
Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10:472–84.
Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–209.
•• Schiavon G, Hrebien S, Garcia-Murillas I, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med. 2015;7:313ra182. Detection of ESR1 mutations in ctDNA is highly sensitive and specific and associated with poor response to subsequent aromatase inhibitor treatment.
•• Fribbens C, O'Leary B, Kilburn L, et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol. 2016;34:2961–8. Retrospective analysis of ESR1 mutations in ctDNA using archival blood samples from a randomized clinical trial.
Chandarlapaty S, Chen D, He W, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016;2:1310–5.
Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13.
Turner NC, Reis-Filho JS. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012;13:e178–85.
Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738–48.
Wardell SE, Nelson ER, Chao CA, McDonnell DP. Bazedoxifene exhibits antiestrogenic activity in animal models of tamoxifen-resistant breast cancer: implications for treatment of advanced disease. Clin Cancer Res. 2013;19:2420–31.
Garner F, Shomali M, Paquin D, et al. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anti-Cancer Drugs. 2015;26:948–56.
Lai A, Kahraman M, Govek S, et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem. 2015;58:4888–904.
Wardell SE, Ellis MJ, Alley HM, et al. Efficacy of SERD/SERM hybrid-CDK4/6 inhibitor combinations in models of endocrine therapy-resistant breast cancer. Clin Cancer Res. 2015;21:5121–30.
Joseph JD, Darimont B, Zhou W et al. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. Elife 2016; 5.
Lonard DM, O'Malley BW. Molecular pathways: targeting steroid receptor coactivators in cancer. Clin Cancer Res 2016.
Acknowledgements
This paper was supported by the NCI Grant K08 CA191058-02 (R.J.), the Breast Cancer Alliance Exceptional Award Grant (R.J.), the Breast Cancer Research Foundation (R.S.), NIH Breast Cancer Specialized Programs of Research Excellence Grants P50CA058183 and P50CA186784 (R.S.), NIH Cancer Center Grant P30CA125123, Susan G. Komen for the Cure Foundation Promise Grants PG12221410 (R.S.), and the Conquer Cancer Foundation (C.D.A., Breast Cancer Research Fellowship).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Rinath Jeselsohn declares no conflict of interest.
Carmine De Angelis declares that he has no conflict of interest.
Myles Brown declares that he has no conflict of interest.
Rachel Schiff has received research support through grants from Gilead and AstraZeneca.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Breast Cancer
Rights and permissions
About this article

Cite this article
Jeselsohn, R., De Angelis, C., Brown, M. et al. The Evolving Role of the Estrogen Receptor Mutations in Endocrine Therapy-Resistant Breast Cancer. Curr Oncol Rep 19, 35 (2017). https://doi.org/10.1007/s11912-017-0591-8
Published:
DOI: https://doi.org/10.1007/s11912-017-0591-8