
Abstract
Alzheimer’s disease (AD) is increasing in prevalence and has a significant impact on caregivers and the healthcare system. One of the many physiologic process affected by AD is the circadian system, with disruption reflected in abnormalities of the sleep-wake cycle. This interaction is bidirectional, with circadian and sleep disruption influencing disease progression. Understanding the bidirectional relationship between AD and circadian disruption may allow for earlier recognition of the potential to develop dementia as well as improved targeted approaches for therapy. Therapies including melatonin and bright light therapy may be advantageous in improving sleep and circadian rhythms and preventing the progression of disease. However, unfortunately, these modalities are not curative, and additional research is needed to improve treatment options for these individuals.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
With the aging of our population, the prevalence of Alzheimer’s disease (AD) and its implications on healthcare and institutionalization increases. AD is currently the most common cause of dementia in the elderly population. In 2013, it was estimated that 4.7 million people between the age of 65 years or older were affected in the USA and this number is expected to reach 14 million by the year 2050 [1, 2]. Circadian rhythm disorders and sleep disturbances occur frequently in AD, with approximately 45% of these patients have reporting problems with sleep. These symptoms may present years prior to the clinical diagnosis of AD [1, 3,4,5]. Additionally, disturbances in the sleep-wake rhythm impact the quality of life of patients and the caregivers of patients in a negative way, resulting in frequent institutionalization of these individuals [6]. Early recognition of sleep and circadian disturbances may provide an early biomarker of AD, while treating these disturbances can serve as a target to improve quality of life, decrease institutionalization in these individuals, and possibly even modify the disease course.
Circadian Rhythms and Sleep
The circadian rhythm is an approximately 24-h rhythm that is reflected in the daily physiologic mechanisms of the body and our behavior. This rhythm is coordinated by the master circadian pacemaker located in the suprachiasmatic nucleus (SCN) in the hypothalamus, which contains two distinct cell types, one containing vasoactive intestinal peptide (VIP) and the other containing vasopressin (AVP) [7,8,9]. The SCN is primarily influenced by light from the retina and by the secretion of melatonin from the pineal gland. The human circadian rhythm is usually slightly longer than 24 h, but is entrained by environmental cues on a daily basis, with adjustments provided by signals or zeitgebers, the two most prominent being light and melatonin [7, 10, 11].
The daily rhythm of the SCN is regulated at a cellular level by a system of transcriptional and translational feedback loops involving clock genes. During the early part of the day, two important transcription factors named BMAL1 and CLOCK dimerize and activate the transcription of the genes PERIOD (PER) and CRYPTOCHROME (CRY). As the day progresses, PER and CRY dimerize to inhibit BMAL1 and CLOCK transcription, therefore limiting their own expression. Later at night, PER and CRY then degrade, thus ending the negative feedback of BMAL1 and CLOCK and allowing for the start of a new circadian cycle. Additionally, BMAL1 and CLOCK also affect the nuclear receptors REV-ERBα and ROR, which inhibit (REV-ERBα) and activate (ROR) the transcription of BMAL1 [12, 13].
The sleep-wake cycle is an important physiological output of the circadian clock and involves several regions of the brain. The ventrolateral preoptic area (VLPO) in the anterior hypothalamus contains inhibitory neurons that are active primarily during sleep [12, 14]. Hypocretin (orexin) neurons in the lateral hypothalamus are active primarily during wakefulness [12, 15, 16]. Additionally, the locus coeruleus (LC), raphe nucleus, pedunculopontine tegmental (PPTg), and laterodorsal tegmental (LDTg) nuclei in the brainstem play an important role in the sleep-wake cycle. Activation of hypocretin neurons induces wakefulness through projections to the LC, LDTg, and PPTg, which leads to arousal [12, 17,18,19,20]. The sleep-promoting VLPO and the wake-promoting nuclei mutually inhibit each other, resulting in the transition from sleep to wake, as per the classically described “flip-flop” model between sleep and wakefulness. Hypocretin serves to stabilize these state transitions [12, 21].
The SCN also sends inhibitory projections to the pineal gland, resulting in a decrease in melatonin release during the day. During the night, when the SCN is inhibited, the release of melatonin from the pineal gland increases, typically starting 2 h before habitual bedtime [7, 22, 23].
The cholinergic system also plays an important role regulating sleep-wake states. Acetylcholine is increased during wake and stage REM sleep and is decreased during NREM sleep. The cholinergic input to the suprachiasmatic nucleus (SCN) originates from the basal forebrain and from the PPTg and LDTg in the brainstem [24]. A recent study by Yamakawa showed that activation of the basal forebrain, either through sleep deprivation or electrical stimulation causes phase advances during the inactive period. These phase shifts are blocked when animals are pretreated with atropine (a cholinergic antagonist) injections to the SCN [25]. Activation of the cholinergic LDTg and PPTg, on the other hand, does not phase shift the circadian clock during the inactive period, but rather causes phase delays when stimulated during the early evening, similar to the phase-shifting effects of light [26].
Sleep and Circadian Disruption in Alzheimer’s Disease
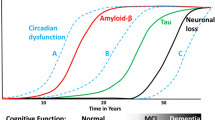
In addition to the hallmark symptom of progressive memory decline, sleep disturbance, disorientation, and agitation are common symptoms in patients with AD, resulting in a significant decline in quality of life. Pathologically, AD is characterized by the combination of amyloid plaques (β-amyloid) and tau containing neurofibrillary tangles. β-Amyloid deposition may play a role in the development and worsening of sleep and circadian symptoms [12, 27].
Circadian dysfunction in AD can manifest in many ways; however, changes to the sleep-wake cycle are the most noticeable. Irregular sleep-wake rhythm disorder, with multiple bouts of sleep within a 24-h period, is frequently observed in patients with neurodegenerative disease. Patients report symptoms of insomnia, with either difficulty falling or staying asleep, and excessive daytime sleepiness [28]. Of note, increased daytime sleepiness has been shown to be associated with an increased risk for dementia [12, 29].
While circadian disruption may be a result of AD, it may in turn lead to worsening of the primary symptoms of memory loss. In the rat, chronic disruption of the circadian rhythm has been shown to impair hippocampal memory. Chronically phase-shifted rats showed deficits in the acquisition and retention of memory when tested using a water maze; however, acutely phase-shifted rats showed no deficits [30]. This suggests that the presence of circadian disruption in AD patients may worsen cognitive decline.
Mechanisms for the Interaction Between Alzheimer’s Disease and Circadian Disruption
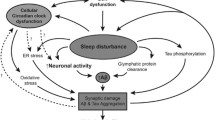
The means by which AD leads to sleep and circadian disruption is likely multifactorial, reflecting pathology at the level of the SCN, as well as inputs and outputs of the circadian clock. In addition, there is evidence that many of these pathways are bidirectional (Fig. 1).

The bidirectional relationship between circadian and sleep-wake disruption and Alzheimer’s disease
Neuronal Loss in the SCN
Autopsy studies of AD patients demonstrate significant loss of neurons in the SCN, correlating with a loss of the amplitude in the circadian rest-activity pattern [12, 31,32,33,34]. More specifically, the AVP-containing neurons and the expression of the MT1 subtype of melatonin receptor are the most severely impacted [7, 31, 32, 35, 36]. As a result, the ability of the SCN to generate a daily rhythm and respond to phase-resetting signals are both impacted.
β-Amyloid Deposition
β-Amyloid has been shown to directly disrupt the output of the circadian clock, often reflected in disruption of the sleep-wake cycle. In rodents, grafting cells that express high levels of β-amyloid into the SCN of adult rats resulted in a significant impairment of rest-activity patterns [37]. In a Drosophila model of AD, it was shown that β-amyloid significantly decreased sleep, mediated by the activation of Jun-N-terminal kinase (JNK). Inhibiting JNK restored normal sleep patterns, decreased β-amyloid accumulation, and decreased further neurodegeneration [38]. In human studies, cognitively normal adults with β-amyloid deposition have been shown to have sleep and circadian disturbances [12, 39]. Additionally, the presence of the APOE ε4 allele, a risk factor for AD, in a healthy population has been shown to result in objective sleep disturbances, using PSG and actigraphy.
There is growing evidence that there is a bidirectional relationship between sleep and the presence of β-amyloid [38, 40]. In patients with preclinical AD, β-amyloid deposition is associated with poor sleep quality [38, 39], while short sleep has been associated with higher β-amyloid deposition in older adults [38, 41]. In mouse models of AD, sleep disruption correlated with β-amyloid plaque formation [38, 42, 43]. β-Amyloid deposition can be present up to two decades prior to the onset of symptomatic AD [38, 44], indicating that sleep and circadian disturbances may serve as an early biomarker for the later progression to AD [38].
Under normal conditions, β-amyloid levels in the brain have a diurnal variation, such that CSF β-amyloid concentrations decrease with sleep and increase with activity [12, 42, 45]. Sleep deprivation leads to elevations in interstitial fluid levels of β-amyloid, in turn resulting in increased formation of plaques, while increasing the total sleep time reduces β-amyloid deposition [12, 43, 46]. These findings are thought to be mediated by neuronal firing. With increased neuronal activity observed during wakefulness, β-amyloid levels increase [47,48,49,50]. Slow-wave sleep, on the other hand, is associated with an overall decrease in neuronal firing and therefore decreased β-amyloid production [47, 51]. Reduced slow-wave sleep, which is seen in AD or sleep deprivation, results in higher levels of neuronal activity and therefore greater β-amyloid production [47, 52, 53, 54]. This reinforces the theory that sleep deprivation increases plaque burden, while increased plaque burden in turn results in increased sleep disruption, in a positive feedback loop.
Recent work by Xie et al. proposed a mechanism by which β-amyloid is cleared from the brain during sleep through the glymphatic system. In mice, sleep results in improved clearance of β-amyloid [55]. As a result, improved sleep may reduce the deposition of β-amyloid within the brain due to more effective clearance during sleep.
Clock Genes
β-Amyloid may also directly affect circadian regulation due to disruptions in circadian clock gene expression. Using an AD mouse model, Song et al. showed that β-amyloid results in degradation of BMAL1 and CBP (CREB-binding protein), causing alterations in the expression of clock genes such as BMAL1 and PER2 and subsequent changes in the circadian rhythm [53].
Circadian clock genes may play a role in the onset and advancement of neurodegeneration. Presenilin-2, a gene that is mutated in hereditary cases of AD, is under clock control. Presenilin-2 normally cleaves amyloid precursor protein (APP), and its expression is activated by the presence of CLOCK and BMAL1 [12, 56, 57]. This implies that circadian disruption and alteration of the transcription/translation feedback loop may also influence the clearance of amyloid.
Melatonin
Melatonin is released from the pineal gland and serves to not only regulate the circadian rhythm but may also prevent further accumulation of β-amyloid. CSF melatonin has been shown to be decreased in patients with AD, even during the preclinical stages [1, 58, 59] and continues to decrease with the progression of disease [1, 58, 60]. Additionally, the ApoE4 allele, a risk factor for AD, has been associated with decreased levels of CSF melatonin [1, 61]. Melatonin itself also regulates the metabolism of APP and directly reduces the levels and prevents the accumulation of β-amyloid [1, 62]. This suggests that the loss of melatonin could lead to further progression of disease secondary to an increase in β-amyloid deposition in the brain.
Cholinergic Disturbance
The cholinergic basal forebrain also undergoes neurodegeneration in AD. Cells of the nucleus basalis magnocellularis, which project to the SCN, can alter circadian rhythms [36, 63]. Erhardt et al. lesioned the cholinergic basal forebrain projections to the SCN in rats, and they found reduced phase advances and increased phase delays in response to light [36, 64]. This suggests that the neurodegeneration associated with AD may in turn be influencing the ability of the circadian system to respond to entraining signals.
Melanopsin-Containing Retinal Ganglion Cell Loss
Melanopsin-expressing retinal ganglion cells (mRGCs) are photoreceptors within the retina that provide the circadian signal of light to the SCN. These cells make up only 1–2% of all RGCs, but transmit information directly to the SCN via the retinohypothalamic tract [65]. When compared to age-matched control patients, AD patients have a greater loss of mRGCs, and the remaining mRGCs exhibited a decreased dendritic diameter and an increase in β-amyloid deposits, suggesting impairment in the few cells that remained [65, 66]. As a result, it is likely that the ability to transmit the circadian signal of light from the retina to the SCN is impaired.
Metabolic Dysfunction
Sleep and circadian disruption have also been linked with metabolic dysfunction, which in itself serves as a risk factor for AD [12, 67,68,69,70,71]. Alterations in the circadian rhythm, including disrupted sleep, reduced sleep duration and irregular sleep patterns can all affect feeding and hormone release patterns, which in turn negatively impact metabolism, resulting in hyperinsulinemia and impaired glucose tolerance [72]. The presence of insulin resistance results in an increased risk of progression to AD and obesity in childhood may also increase the risk of cognitive impairment with aging [12, 73, 74].
Treatment
The association between sleep and circadian rhythm disruption and the progression of AD presents an opportunity to target preclinical symptoms of AD to prevent further progression of neurodegeneration. Focusing on early treatment may limit institutionalization and healthcare burden. Potential treatments including bright light therapy, melatonin, and other pharmacologic treatments may serve to diminish the prevalence or severity of AD.
Non-pharmacologic Treatments
Irregular daily rest-activity and behavioral patterns are common in patients with AD. In order to restore regularity to the daily routine, establishing proper sleep hygiene techniques is important, especially in elderly institutionalized patients with limited light exposure. A randomized trial of nursing home patients showed that the combination of increased daytime physical activity and an improved nighttime sleeping environment, with decreased noise and limited nursing interventions resulted in an improvement in overall sleep and agitation [75]. Patients with AD should be encouraged to exercise regularly and maintain a regular sleep-wake schedule. Daytime naps should be limited and noise and light exposure should be limited at night.
Bright light therapy (BLT) is frequently administered to treat circadian rhythm disturbances and may be beneficial for addressing the circadian dysregulation seen in patients with AD. Ten days of evening BLT (3000 lx) for 2 h improved cognitive functioning of patients with dementia, assessed using mini-mental status examination scores [76]. In another randomized controlled trial, 4 weeks of morning bright light therapy (10,000 lx) for 1 h per day resulted in improvements of sleep from an average of 6.4 h per night to 8.1 h per night [77]. Another randomized trial of nursing home residents with AD demonstrated that 10 weeks of exposure to bright light therapy (≥2500 lx, 1 h) resulted in improved agitation, depression, and appetite [78].
A recent systematic review and meta-analysis looking at the effects of light therapy in circadian rhythm sleep disorders, insomnia, AD, and dementia found small to medium, but significant effects of light therapy on outcomes in patients with AD [79•]. Significant improvements with BLT were seen for sleep onset latency, total sleep time, time in bed, sleep efficiency, and sleep quality, but not for wake after sleep onset and early morning awakenings [79•]. A Cochrane review looking at light therapy and its effects on cognition, sleep, and behavior in patients with dementia showed inconclusive results, however. Eleven randomized controlled trials were assessed and found no effect of light therapy on cognitive function, sleep, or agitation associated with dementia. However, this review was not limited to AD and included other types of dementia, which may have limited these findings [80].
There is currently no consensus regarding the effective duration and intensity of bright light therapy for the treatment of sleep and circadian disruption in AD due to the heterogeneity and small sample size of most research studies. The disparities seen in current results may be due to the substantial variability in the clinical populations, including the type of dementia, severity of disease, and degree of visual impairment. In addition, baseline environmental light exposure and the duration, intensity, and time of light intervention were quite varied. Nonetheless, BLT appears to be a potentially effective and low-cost intervention with minimal side effects.
Pharmacologic Treatments
Melatonin, another circadian zeitgeber, may serve as an additional therapeutic agent for treating circadian rhythm disruption in patients with AD. Current research has focused on the effects of melatonin on sleep, cognition, and AD progression. In a study using a rat model of AD (OXYS rats), melatonin was shown to decrease β-amyloid levels within the hippocampus and frontal cortex. Additionally, when started in the early stages of AD progression, melatonin was shown to slow hippocampal degeneration, especially in the CA1 region. Melatonin administration may therefore decrease the progression of AD pathology and may serve as an important intervention to limit further β-amyloid deposition [81].
In a small double-blind study, melatonin (3 mg), given at 8:30 p.m. daily for 4 weeks, resulted in prolonged sleep time and decreased nighttime activity [82]. One of the longest duration randomized placebo -trials over a 6-month period looked the effect of prolonged release melatonin (2 mg) on sleep and cognitive function in 80 patients with mild to moderate AD. Patients treated with melatonin had improved cognition and sleep efficiency, with minimal side effects [83]. A recent meta-analysis of the use of melatonin in AD showed that patients receiving melatonin had a prolonged total sleep time, however no improvement in cognitive abilities. Doses of melatonin ranged from 1.5 to 10 mg with a duration of treatment ranging from 10 days to 24 weeks [84•]. There is some concern that melatonin and melatonin agonists may be less effective in late clinical AD patients, as the number and density of melatonin MT1 receptors in the SCN is decreased in later stages of AD [85]. Given the variability of doses, duration of treatment, severity of dementia, and additional comorbidities in these patients, it is difficult to make practice recommendations based on these results, but melatonin does appear to benefit nighttime total sleep time.
There is also evidence for the benefit of melatonin in conjunction with BLT. One study showed that the combination of melatonin (5 mg) and bright light treatment (≥2500 lx for 1 h for 5 days per week) over a 10-week period, resulted in a reduced daytime sleep, increased daytime activity and improved day versus night sleep ratio. In this study, BLT alone did not improve nighttime sleep, daytime sleepiness, or rest-activity rhythms [86]. The combination with light therapy and melatonin may be a better approach by targeting multiple routes for circadian entrainment, as it is known that both retinal light input pathways and melatonin receptor density at the SCN may be affected by disease progression.
Alternatives to melatonin could include off-label use of the prescription melatonin M1/M2 receptor agonists, ramelteon and tasimelteon. While tasimelteon has not been studied in AD patients, in a case series ramelteon was used to successfully treat delirium in five patients. Possible mechanisms include improving the underlying circadian rhythm disruption, though further research is needed to better assess its efficacy [87].
Looking at other prescription hypnotics, one study evaluated the use of trazodone (50 mg) at bedtime. Patients showed improved total sleep time at night and no significant adverse side effects [88, 89]. In another study, actigraphy data in 30 AD patients before and after trazodone use was recorded for 2 weeks. Results demonstrated a significant improvement in relative rhythm amplitude, compatible with a more stable daytime behavioral pattern, indicating that trazodone may stabilize circadian rhythms in individuals with AD [90], though more research trials need to be done to demonstrate significant benefit.
Conclusion
Circadian dysfunction is a pervasive problem in AD patients, which may occur years prior to clinical AD onset and increase with severity of disease due to a bidirectional relationship. Given the increased behavioral complications and the frequent need for institutionalization due to high caretaker burden, further work in assessing the effectiveness of chronotherapeutic agents in the treatment of AD is warranted. Detecting early symptoms of AD, with changes in sleep patterns and irregularities in rest-activity rhythms may provide early biomarkers for intervention to prevent further β-amyloid deposition and potentially slow the progression of neurodegenerative disease. In addition, added benefits would include improving sleep, limiting behavioral disruptions and improving the quality of life of patients with early dementia and their caregivers.
Further research into the underlying pathology of AD and the link with circadian disruption is warranted. Additionally, more research trials assessing effective treatment options are needed to better address circadian dysfunction in patients with AD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Urrestarazu E, Iriarte J. Clinical management of sleep disturbances in Alzheimer’s disease: current and emerging strategies. Nature and science of sleep. 2016;8:21–33.
Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013;80:1778–83.
Pistacchi M, Gioulis M, Contin F, Sanson F, Marsala SZ. Sleep disturbance and cognitive disorder: epidemiological analysis in a cohort of 263 patients. Neurological Sciences: official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2014;35:1955–62.
Moran M, Lynch CA, Walsh C, Coen R, Coakley D, Lawlor BA. Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med. 2005;6:347–52.
Cipriani G, Lucetti C, Danti S, Nuti A. Sleep disturbances and dementia. Psychogeriatrics: the official journal of the Japanese Psychogeriatric Society. 2015;15:65–74.
Hope T, Keene J, Gedling K, Fairburn CG, Jacoby R. Predictors of institutionalization for people with dementia living at home with a carer. International Journal of Geriatric Psychiatry. 1998;13:682–90.
Slats D, Claassen JA, Verbeek MM, Overeem S. Reciprocal interactions between sleep, circadian rhythms and Alzheimer’s disease: focus on the role of hypocretin and melatonin. Ageing Res Rev. 2013;12:188–200.
Jin X, Shearman LP, Weaver DR, Zylka MJ, de Vries GJ, Reppert SM. A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell. 1999;96:57–68.
Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–41.
Cassone VM, Chesworth MJ, Armstrong SM. Entrainment of rat circadian rhythms by daily injection of melatonin depends upon the hypothalamic suprachiasmatic nuclei. Physiol Behav. 1986;36:1111–21.
Johnson RF, Moore RY, Morin LP. Loss of entrainment and anatomical plasticity after lesions of the hamster retinohypothalamic tract. Brain Res. 1988;460:297–313.
Mattis J, Sehgal A. Circadian rhythms, sleep, and disorders of aging. Trends in Endocrinology and Metabolism: TEM. 2016;27:192–203.
Lowrey PL, Takahashi JS. Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004;5:407–41.
Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science (New York, NY). 1996;271:216–9.
de Lecea L, Kilduff TS, Peyron C, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–7.
Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci Off J Soc Neurosci. 2005;25:6716–20.
Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–4.
Carter ME, Adamantidis A, Ohtsu H, Deisseroth K, de Lecea L. Sleep homeostasis modulates hypocretin-mediated sleep-to-wake transitions. J Neurosci Off J Soc Neurosci. 2009;29:10939–49.
Carter ME, Yizhar O, Chikahisa S, et al. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci. 2010;13:1526–33.
Peyron C, Tighe DK, van den Pol AN, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci Off J Soc Neurosci. 1998;18:9996–10015.
Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63.
Gerdin MJ, Masana MI, Rivera-Bermudez MA, et al. Melatonin desensitizes endogenous MT2 melatonin receptors in the rat suprachiasmatic nucleus: relevance for defining the periods of sensitivity of the mammalian circadian clock to melatonin. FASEB Journal: official publication of the Federation of American Societies for Experimental Biology. 2004;18:1646–56.
Tzischinsky O, Shlitner A, Lavie P. The association between the nocturnal sleep gate and nocturnal onset of urinary 6-sulfatoxymelatonin. J Biol Rhythm. 1993;8:199–209.
Hut RA, Van der Zee EA. The cholinergic system, circadian rhythmicity, and time memory. Behav Brain Res. 2011;221:466–80.
Yamakawa GR, Basu P, Cortese F, et al. The cholinergic forebrain arousal system acts directly on the circadian pacemaker. Proc Natl Acad Sci U S A. 2016;113:13498–503.
Abbott SM, Arnold JM, Chang Q, et al. Signals from the brainstem sleep/wake centers regulate behavioral timing via the circadian clock. PLoS One. 2013;8:e70481.
Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA neurology. 2014;71:505–8.
Abbott SM, Zee PC. Irregular sleep-wake rhythm disorder. Sleep medicine clinics. 2015;10:517–22.
Merlino G, Piani A, Gigli GL, et al. Daytime sleepiness is associated with dementia and cognitive decline in older Italian adults: a population-based study. Sleep Med. 2010;11:372–7.
Craig LA, McDonald RJ. Chronic disruption of circadian rhythms impairs hippocampal memory in the rat. Brain Res Bull. 2008;76:141–51.
Stopa EG, Volicer L, Kuo-Leblanc V, et al. Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J Neuropathol Exp Neurol. 1999;58:29–39.
Zhou JN, Hofman MA, Swaab DF. VIP neurons in the human SCN in relation to sex, age, and Alzheimer’s disease. Neurobiol Aging. 1995;16:571–6.
Swaab DF, Fliers E, Partiman TS. The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 1985;342:37–44.
Wang JL, Lim AS, Chiang WY, et al. Suprachiasmatic neuron numbers and rest-activity circadian rhythms in older humans. Ann Neurol. 2015;78:317–22.
Liu RY, Zhou JN, Hoogendijk WJ, et al. Decreased vasopressin gene expression in the biological clock of Alzheimer disease patients with and without depression. J Neuropathol Exp Neurol. 2000;59:314–22.
Coogan AN, Schutova B, Husung S, et al. The circadian system in Alzheimer’s disease: disturbances, mechanisms, and opportunities. Biol Psychiatry. 2013;74:333–9.
Tate B, Aboody-Guterman KS, Morris AM, Walcott EC, Majocha RE, Marotta CA. Disruption of circadian regulation by brain grafts that overexpress Alzheimer beta/A4 amyloid. Proc Natl Acad Sci U S A. 1992;89:7090–4.
Song Q, Feng G, Huang Z, Chen X, Chen Z, Ping Y. Aberrant axonal arborization of PDF neurons induced by Abeta42-mediated JNK activation underlies sleep disturbance in an Alzheimer’s model. Mol Neurobiol. 2016.
Ju YE, McLeland JS, Toedebusch CD, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurology. 2013;70:587–93.
Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology—a bidirectional relationship. Nat Rev Neurol. 2014;10:115–9.
Spira AP, Gamaldo AA, An Y, et al. Self-reported sleep and beta-amyloid deposition in community-dwelling older adults. JAMA Neurology. 2013;70:1537–43.
Roh JH, Huang Y, Bero AW, et al. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med. 2012;4:150ra122.
Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cyclee. Science (New York, NY) 2009; 326:1005–1007.
Selkoe DJ. The therapeutics of Alzheimer’s disease: where we stand and where we are heading. Ann Neurol. 2013;74:328–36.
Huang Y, Potter R, Sigurdson W, et al. Effects of age and amyloid deposition on Abeta dynamics in the human central nervous system. Arch Neurol. 2012;69:51–8.
Tabuchi M, Lone SR, Liu S, et al. Sleep interacts with abeta to modulate intrinsic neuronal excitability. Current biology: CB. 2015;25:702–12.
Musiek ES, Xiong DD, Holtzman DM. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp Mol Med. 2015;47:e148.
Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron. 2003;37:925–37.
Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–22.
Bero AW, Yan P, Roh JH, et al. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci. 2011;14:750–6.
Nir Y, Staba RJ, Andrillon T, et al. Regional slow waves and spindles in human sleep. Neuron. 2011;70:153–69.
Martin PR, Loewenstein RJ, Kaye WH, Ebert MH, Weingartner H, Gillin JC. Sleep EEG in Korsakoff's psychosis and Alzheimer’s disease. Neurology. 1986;36:411–4.
Song H, Moon M, Choe HK, et al. Abeta-induced degradation of BMAL1 and CBP leads to circadian rhythm disruption in Alzheimer’s disease. Mol Neurodegener. 2015;10:13.
Varga AW, Wohlleber ME, Gimenez S, et al. Reduced slow-wave sleep is associated with high cerebrospinal fluid Abeta42 levels in cognitively normal elderly. Sleep. 2016;39:2041–8.
Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science (New York, NY). 2013;342:373–7.
Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science (New York, NY). 1995;269:973–7.
Belanger V, Picard N, Cermakian N. The circadian regulation of Presenilin-2 gene expression. Chronobiol Int. 2006;23:747–66.
Mishima K, Tozawa T, Satoh K, Matsumoto Y, Hishikawa Y, Okawa M. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol Psychiatry. 1999;45:417–21.
Wu YH, Swaab DF. The human pineal gland and melatonin in aging and Alzheimer’s disease. J Pineal Res. 2005;38:145–52.
Zhou JN, Liu RY, Kamphorst W, Hofman MA, Swaab DF. Early neuropathological Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res. 2003;35:125–30.
Liu RY, Zhou JN, van Heerikhuize J, Hofman MA, Swaab DF. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-epsilon 4/4 genotype. J Clin Endocrinol Metab. 1999;84:323–7.
Lin L, Huang QX, Yang SS, Chu J, Wang JZ, Tian Q. Melatonin in Alzheimer’s disease. Int J Mol Sci. 2013;14:14575–93.
Colwell CS, Kaufman CM, Menaker M. Phase-shifting mechanisms in the mammalian circadian system: new light on the carbachol paradox. J Neurosci Off J Soc Neurosci. 1993;13:1454–9.
Erhardt C, Galani R, Jeltsch H, et al. Modulation of photic resetting in rats by lesions of projections to the suprachiasmatic nuclei expressing p75 neurotrophin receptor. Eur J Neurosci. 2004;19:1773–88.
Feng R, Li L, Yu H, Liu M, Zhao W. Melanopsin retinal ganglion cell loss and circadian dysfunction in Alzheimer’s disease (review). Mol Med Rep. 2016;13:3397–400.
La Morgia C, Ross-Cisneros FN, Koronyo Y, et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann Neurol. 2016;79:90–109.
Dibner C, Schibler U. Circadian timing of metabolism in animal models and humans. J Intern Med. 2015;277:513–27.
Maury E, Hong HK, Bass J. Circadian disruption in the pathogenesis of metabolic syndrome. Diabetes & Metabolism. 2014;40:338–46.
Weljie AM, Meerlo P, Goel N, et al. Oxalic acid and diacylglycerol 36:3 are cross-species markers of sleep debt. Proc Natl Acad Sci U S A. 2015;112:2569–74.
Davies SK, Ang JE, Revell VL, et al. Effect of sleep deprivation on the human metabolome. Proc Natl Acad Sci U S A. 2014;111:10761–6.
Taheri S, Lin L, Austin D, Young T, Mignot E. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med. 2004;1:e62.
Forrestel AC, Miedlich SU, Yurcheshen M, Wittlin SD, Sellix MT. Chronomedicine and type 2 diabetes: shining some light on melatonin. Diabetologia. 2016.
Willette AA, Bendlin BB, Starks EJ, et al. Association of insulin resistance with cerebral glucose uptake in late middle-aged adults at risk for Alzheimer disease. JAMA neurology. 2015;72:1013–20.
Luciano R, Barraco GM, Muraca M, et al. Biomarkers of Alzheimer disease, insulin resistance, and obesity in childhood. Pediatrics. 2015;135:1074–81.
Alessi CA, Yoon EJ, Schnelle JF, Al-Samarrai NR, Cruise PA. A randomized trial of a combined physical activity and environmental intervention in nursing home residents: do sleep and agitation improve? J Am Geriatr Soc. 1999;47:784–91.
Graf A, Wallner C, Schubert V, et al. The effects of light therapy on mini-mental state examination scores in demented patients. Biol Psychiatry. 2001;50:725–7.
Lyketsos CG, Lindell Veiel L, Baker A, Steele C. A randomized, controlled trial of bright light therapy for agitated behaviors in dementia patients residing in long-term care. International Journal of Geriatric Psychiatry. 1999;14:520–5.
Dowling GA, Graf CL, Hubbard EM, Luxenberg JS. Light treatment for neuropsychiatric behaviors in Alzheimer’s disease. West J Nurs Res. 2007;29:961–75.
• van Maanen A, Meijer AM, van der Heijden KB, Oort FJ. The effects of light therapy on sleep problems: a systematic review and meta-analysis. Sleep Med Rev. 2016;29:52–62. A recent systematic review and meta-analysis looking at the effects of light therapy in circadian rhythm sleep disorders, insomnia, AD, and dementia. Eleven studies included patients with AD/dementia and a total of N = 211 patients, demonstrated significant effects of light therapy in patients with AD/dementia in improving sleep onset latency, total sleep time, time in bed, sleep efficiency, and sleep quality. However, there was no significant improvement seen in wake after sleep onset or early morning awakenings.
Forbes D, Blake CM, Thiessen EJ, Peacock S, Hawranik P. Light therapy for improving cognition, activities of daily living, sleep, challenging behaviour, and psychiatric disturbances in dementia. Cochrane Database Syst Rev. 2014: Cd003946
Rudnitskaya EA, Muraleva NA, Maksimova KY, Kiseleva E, Kolosova NG, Stefanova NA. Melatonin attenuates memory impairment, amyloid-beta accumulation, and neurodegeneration in a rat model of sporadic Alzheimer’s disease. Journal of Alzheimer’s Disease: JAD. 2015;47:103–16.
Asayama K, Yamadera H, Ito T, Suzuki H, Kudo Y, Endo S. Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non-cognitive functions in Alzheimer type dementia. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2003;70:334–41.
Wade AG, Farmer M, Harari G, et al. Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: a 6-month, randomized, placebo-controlled, multicenter trial. Clin Interv Aging. 2014;9:947–61.
• Wang YY, Zheng W, Ng CH, Ungvari GS, Wei W, Xiang YT. Meta-analysis of randomized, double-blind, placebo-controlled trials of melatonin in Alzheimer’s disease. International Journal of Geriatric Psychiatry. 2017;32:50–7. A recent meta-analysis looking at the effects of melatonin in AD. Seven studies included patients with AD and a total of N = 462 patients, with the administration of melatonin dosage ranging from 1.5 to 10 mg, from 10 days to 24 weeks, demonstrated that patients receiving melatonin treatment showed prolonged total sleep time at night. Melatonin, however, did not improve cognitive abilities assessed by the mini-mental state examination (MMSE) or the Alzheimer’s Disease Assessment Cognitive Subscale (ADACS).
Wu YH, Zhou JN, Van Heerikhuize J, Jockers R, Swaab DF. Decreased MT1 melatonin receptor expression in the suprachiasmatic nucleus in aging and Alzheimer’s disease. Neurobiol Aging. 2007;28:1239–47.
Dowling GA, Burr RL, Van Someren EJ, et al. Melatonin and bright-light treatment for rest-activity disruption in institutionalized patients with Alzheimer’s disease. J Am Geriatr Soc. 2008;56:239–46.
Furuya M, Miyaoka T, Yasuda H, et al. Marked improvement in delirium with ramelteon: five case reports. Psychogeriatrics: the official journal of the Japanese Psychogeriatric Society. 2012;12:259–62.
Ooms S, Ju YE. Treatment of sleep disorders in dementia. Curr Treat Options Neurol. 2016;18:40.
Camargos EF, Louzada LL, Quintas JL, Naves JO, Louzada FM, Nobrega OT. Trazodone improves sleep parameters in Alzheimer disease patients: a randomized, double-blind, and placebo-controlled study. The American Journal of Geriatric Psychiatry: official journal of the American Association for Geriatric Psychiatry. 2014;22:1565–74.
Grippe TC, Goncalves BS, Louzada LL, et al. Circadian rhythm in Alzheimer disease after trazodone use. Chronobiol Int. 2015;32:1311–4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Yumna Saeed and Sabra M. Abbott declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Sleep
Rights and permissions
About this article

Cite this article
Saeed, Y., Abbott, S.M... Circadian Disruption Associated with Alzheimer’s Disease. Curr Neurol Neurosci Rep 17, 29 (2017). https://doi.org/10.1007/s11910-017-0745-y
Published:
DOI: https://doi.org/10.1007/s11910-017-0745-y