
Abstract
Mainly due to the advent of next-generation sequencing (NGS), the field of genetics of dystonia has rapidly grown in recent years, which led to the discovery of a number of novel dystonia genes and the development of a new classification and nomenclature for inherited dystonias. In addition, new findings from both in vivo and in vitro studies have been published on the role of previously known dystonia genes, extending our understanding of the pathophysiology of dystonia. We here review the current knowledge and recent findings in the known genes for isolated dystonia TOR1A, THAP1, and GNAL as well as for the combined dystonias due to mutations in GCH1, ATP1A3, and SGCE. We present confirmatory evidence for a role of dystonia genes that had not yet been unequivocally established including PRKRA, TUBB4A, ANO3, and TAF1. We finally discuss selected novel genes for dystonia such as KMT2B and VAC14 along with the challenges for gene identification in the NGS era and the translational importance of dystonia genetics in clinical practice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The term “dystonia” was first coined by Hermann Oppenheim in his landmark article entitled “About a peculiar cramping sickness in children and adolescents (dysbasia lordotica progressiva, dystonia musculorum deformans)” [1]. Intriguingly, despite the absence of an obvious family history, Oppenheim suspected that “hereditary burden likely plays a major role” in the etiology of the disease and thus his paper may also represent the first reference to the role of genetics in dystonia [1, 2•].
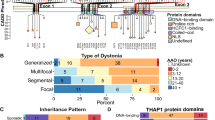
Dystonia is a rare movement disorder with an estimated prevalence of 16:100,000 [3]. It is characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both [4•]. Dystonia is a clinically and genetically highly heterogeneous disorder. Dystonic symptoms can present from early infancy to late adulthood and affect one (focal), several (multifocal and segmental), or many (generalized) body parts. Adult-onset focal dystonia is by far the most frequent form of dystonia. In two recent studies of 1500 Chinese and 600 Irish patients with this type of dystonia, respectively, the vast majority of patients had dystonia of the neck (cervical dystonia, 37 [5] vs. 69% [6]) or the eye lid (blepharospasm, 56 [5] vs. 17% [6]). The differences for the prevalence of blepharospasm may be best explained by the exposure to environmental factors such as sun light [7] or air pollution that directly interact with the eyes. Other types of dystonia such as focal hand or leg dystonia (3–7%), spasmodic dysphonia 18 (1–3%), musician’s dystonia (3%), or oromandibular dystonia (1%) were much rarer [5, 6]. Women are affected about twice as often as men.
Of note, although the genetic causes of dystonia—especially for adult-onset focal forms—are largely elusive, a positive family history has been reported in about 20% of the patients [6, 8, 9]. In the past three decades, more than 200 genes have been linked to different, mainly childhood-onset generalized forms of dystonia (www.omim.org) [10, 11•]. This includes forms in which dystonia is the only disease manifestation with the exception of tremor (“isolated dystonia”), forms in which dystonia co-occurs with another movement disorder such as parkinsonism or myoclonus (“combined dystonia”) and disorders in which dystonia is often less prominent and usually one of several disease manifestations (“complex dystonia”) [4•, 12••]. Notably, most of the genetic forms belong to the latter phenotypic group which, however, also represents the most heterogeneous class in terms of clinical expression.
The genetic nomenclature (DYTs) was originally designed to consecutively number findings from linkage studies in large dystonia families without knowing the underlining gene mutation. Meanwhile, many of the locus symbols were repeatedly confirmed and respective genes with disease-causing mutations identified, e.g., mutations in the TOR1A gene at the DYT1 locus as a cause of autosomal dominant torsion dystonia [13]. The genetic locus symbols (e.g., DYT1) were later also used as synonyms for the respective phenotype such as “DYT1 dystonia” or the underlying genetic cause (mutation in the “DYT1” gene) which is, however, not recommended by the official HUGO Gene Nomenclature Committee (www.genenames.org). Given the rapidly evolving field of (dystonia) genetics, it has become increasingly impossible to maintain a chronological, numbered system and several problems arose, including erroneous or duplicated loci [14]. Therefore, a new genetic classification and nomenclature scheme based on the gene, mutations which have been confirmed as a genetic cause of dystonia, has been proposed by a dedicated Task Force for the Classification and Nomenclature of Genetic Movement Disorders installed by the International Parkinson’s Disease and Movement Disorder Society. According to the Task Force’s recommendations the phenotype prefix “DYT” for dystonia is followed by the gene name [12••]. In the case of DYT1, this would, for example, be replaced by DYT-TOR1A.
Next-generation sequencing (NGS) has led to the description of many new dystonia genes [15]. However, several of these findings need to be interpreted with caution. In this article, we will highlight new findings on several of the established genes for isolated and combined forms of dystonia, include selected examples of novel dystonia genes identified in 2015 and 2016, and provide guidelines for distinguishing false-positive from true-positive genetic findings in the era of NGS.
Current Knowledge and New Findings in “Old” Genes
In this paragraph, we will summarize current knowledge on the three unequivocally established genes for isolated dystonia including TOR1A [13], THAP1 [16], and GNAL [17] (Table 1). These genetic forms are all inherited in an autosomal dominant fashion and found in <5% of dystonia patients. This will be followed by a description of the well-known genes for combined dystonia including GCH1 [18], ATP1A3 [19], and SGCE [20] (Table 1). For a detailed overview on long-known genetic forms of complex dystonia, we would like to refer the reader to another review article [21].
DYT-TOR1A
Mutations in the TOR1A gene (torsin family 1 member A) encoding TorsinA, a member of the AAA+ superfamily (ATPases associated with a variety of cellular activities), are a cause of early-onset generalized dystonia. The first and so far the only clearly established mutation is a 3-bp deletion in the TOR1A gene (c.904_906delGAG; p.302delGlu) [13]. This mutation is frequently found among Ashkenazi Jewish patients due to a founder effect [22]. Different rare missense and in-frame mutations have not only repeatedly been reported in individual patients [23, 24] but also control and lack unequivocal confirmation of pathogenicity [25], although some functions of TorsinA may be impaired in cellular models [24, 26].
DYT-TOR1A dystonia usually presents as dystonia in an extremity in childhood. The symptoms later progress to other body parts but typically spare the face and neck [22]. There is variable expressivity ranging from severe childhood-onset generalized to late-onset focal dystonia and about two thirds of the mutation carriers remain unaffected throughout their life (reduced penetrance) [27]. Deep brain stimulation has repeatedly shown to be an effective treatment option [28•].
Despite linking a mutation in TOR1A to dystonia 20 years ago, the role of TorsinA is still largely elusive. TorsinA is mainly located in the endoplasmatic reticulum and the perinuclear space and, due to its function as a triple AAA+ protein, is thought to act as a molecular chaperone [29]. It is known that TorsinA needs to bind to LAP1 (lamina-associated protein) or LULL1 (luminal domain like LAP1) for activation [30]. This binding is impaired by the GAG deletion [31] as was confirmed by crystallography [32]. It was recently shown that TorsinA is associated with membrane expansion at the inner nuclear membrane and with elevated cellular lipid content suggesting torsins as essential regulators of cellular lipid metabolism. Thus, disturbed lipid biology may be a main contributor to the development of dystonia due to TOR1A mutation(s) [33••].
There is an ongoing debate where the anatomical cause of dystonia is located with a main focus on brainstem and increasingly on cerebellum [34•]. Investigations of TOR1A mutation carriers and respective animal models are used to further explore this issue. For example, a resting-state functional MRI study in symptomatic and asymptomatic mutation carriers, as well as healthy controls points to a crucial role of nodes in sensory-motor integration of posterior parietal structures [35]. Results from different TOR1A mouse models imply altered maturation of cerebellar synaptic contacts [36].
DYT-THAP1
Mutations in the THAP1 gene (THAP domain containing, apoptosis-associated protein 1) encoding the transcription factor THAP1 are a cause of adolescent-onset dystonia with mixed phenotype (previously referred to as DYT6) [16]. About 100 different mutations have been reported in THAP1 including missense, nonsense, and frameshift mutations [37, 38]. DYT-THAP1 dystonia usually presents with dysphonia or writer’s cramp in late childhood or adolescence. Over the course of the disease, dystonia spreads to other body parts with prominent craniocervical involvement [39]. As for DYT-TOR1A, the penetrance of DYT-THAP1 is highly reduced (to about 50%) and there is variable expressivity. As in most other dystonia forms, there seems to be no neurodegeneration and no specific disease-related pathology in DYT-THAP1 [40]. Also for this form of dystonia, deep brain stimulation should be considered as a therapeutic option despite an overall less favorable and less consistent response [41].
THAP1 has a DNA-binding THAP domain at the N-terminus as well as a nuclear localization signal (NLS) and several protein-protein interaction motifs towards the C-terminus. Due to posttranslational modification, several THAP1 species exist including a neuron-specific form that may be a key player in controlling neuronal gene transcription [42]. THAP1 regulates its own expression and represses the expression of TOR1A in vitro while several THAP1 mutations disrupt this repression [43, 44]. Additional targets of the transcription factor THAP1 remain to be identified. The transcriptional control is mediated by transcriptional cofactors such as HCFC1 (host cell factor C1, unpublished data).
DYT-GNAL
Heterozygous mutations in the GNAL gene (guanine nucleotide-binding protein subunit alpha L) encoding the stimulatory α subunit of the heterotrimeric G protein Golf (Gαolf) cause cervical or cranial dystonia with a mean onset in the thirties [17]. About 30 different GNAL mutations have been reported in dystonia patients. In a recent screening study of 1,000 patients with cervical dystonia, five carriers of pathogenic or likely pathogenic mutations were identified including four missense and a nonsense variant [45]. In the same study, an enrichment of the synonymous substitution c.1014C.T (p.A338=) was found among patients compared to samples from the exome aggregation consortium (ExAC) [45]. GNAL mutations seem to be highly but not fully penetrant [46]. Recently, a first homozygous GNAL missense mutation (p.R329W) was found in two sisters with childhood-onset generalized dystonia and mild intellectual disability; the parents were presumably healthy [47•]. Thus, biallelic mutations in GNAL also occur but appear to be related to a more severe phenotype, as is also the case for biallelic mutations in GCH1 [48]. Functional characterization of the homozygous GNAL mutation revealed impaired Gαolf functional coupling to dopamine D1 receptors [47•], while previously described GNAL mutations pointed to a strict loss-of-function mechanism [49].
Gαolf is enriched in the striatum where it couples D1 dopamine (D1R) and A2A adenosine (A2AR) receptors to downstream effector molecules. Of note, coupling to A2AR leads to activation of adenylyl cyclase type 5 (AC5) encoded by ADCY5 [47•], mutations in which are another cause of dystonia and additional movement disorders (see below) [50•].
DYT-GCH1
Heterozygous mutations in the GCH1 gene (GTP cyclohydrolase 1), encoding the rate-limiting enzyme in the biosynthesis of dopamine via the biopterin pathway, are a cause of childhood-onset dopa-responsive dystonia (DRD) with diurnal fluctuation [18]. Parkinsonian features often co-occur or may be the only finding [51, 52] and even be associated with a presynaptic dopaminergic deficit as evidenced by SPECT [53]. Further, GCH1 variants may also represent a risk factor for PD [54]. To date, more than 100 different mutations have been reported including missense, nonsense, frameshift, and splice-site mutations all over the gene, as well as whole-exon or whole-gene deletions [51, 55]. GCH1 mutation carriers show a penetrance of around 50% which is considerably higher in women compared to men. Recessively inherited (biallelic) mutations in GCH1 result in a much more severe clinical phenotype including developmental delay and infantile onset [48].
Due to the enzymatic defect in the levodopa biosynthesis, there is a life-long response of DRD to levodopa therapy. Dopamine signaling is important already for intra-uterine (brain) development [56], and prenatal treatment with levodopa in a homozygous GCH1 mutation carrier was shown to prevent development of the severe phenotype related to biallelic GCH1 mutations [57]. Of note, biopterin is not only an important intermediate substrate for the biosynthesis of dopamine but also of other neurotransmitters including serotonin. This may explain why non-motor features such as sleep disturbances, mood disorders, or migraine are frequent among patients with DRD [51].
DYT-ATP1A3
Mutations in the ATP1A3 gene (ATPase Na+/K+ transporting subunit alpha 3), encoding an ionic pump, are a cause of rapid-onset dystonia-parkinsonism (RDP) with a characteristic and as the name indicates, sudden onset usually in adolescence or young adulthood [19]. The acute onset is usually associated with a trigger such as fever, physical exertion, or emotional stress [58]. Dystonic symptoms frequently show a rostro-caudal gradient with a strong involvement of the bulbar region and are often accompanied by bradykinesia as a parkinsonian feature [58]. Symptoms usually only slightly respond to levodopa but to a certain degree to benzodiazepine and clonazepam therapy. RDP is inherited in an autosomal dominant manner with reduced penetrance. In addition, mutations in ATP1A3 have also been linked to a variety of clinical syndromes (pleiotropy) including epileptic or hemiplegic attacks, ataxia, cognitive decline, and other neurological disorders, often with a more severe course and an earlier age at onset [59•].
Proper function of the ionic pump ATP1A3 that uses ATP hydrolysis to exchange Na+/K+ ions is important to maintain an electrochemical gradient across the plasma membrane that is a prerequisite for electrical excitability and secondary transport in neurons. In a mouse knock-in model, it was recently shown that this ATPase is involved in the control of spatial learning and memory and may be linked to GABA transmission [60].
About 20 different missense mutations were identified in ATP1A3. In about 50% of the patients, ATP1A3 mutations arose de novo or were inherited from parents with mosaics [61]. Most of the mutations are unique but a mutational hot spot seems to be located at protein position D801 [60]. It has been demonstrated that RDP-causing mutations reduce protein levels of ATP1A3 but do not affect the enzymatic activity per se [19].
DYT-SGCE
Mutations in the SGCE gene (sarcoglycan epsilon), coding for the ε member of the sarcoglycan family, are a frequent cause of myoclonus-dystonia (M-D) characterized by predominant, action-induced, alcohol-responsive myoclonic jerks [20, 62]. Myoclonus as well as dystonia most commonly involves the neck or arms/hands. Onset is usually in childhood or adolescence [63]. Additionally, many carriers of SGCE mutations develop psychiatric features such as anxiety-related disorders and alcohol dependence, which is important for counseling and specific treatment [64]. About 80 different mutations have been reported in SGCE, many of which are loss-of-function mutations due to frameshift, splice site, nonsense mutations, or deletions of whole exons or even the entire gene. The latter type of mutation often also involves loss of adjacent genes leading to additional clinical features such as joint problems [62].
As for all autosomal dominantly inherited isolated and combined dystonias, M-D also shows reduced penetrance—but in contrast to the other forms—for SGCE, the underlying mechanism has been elucidated: penetrance of SGCE mutations is only reduced upon maternal transmission due to maternal genomic imprinting of the SGCE gene [65]. This impacts on genetic counseling since it is very unlikely that offspring who inherited the mutation from the mother will develop the disease because only the wildtype allele from the father will be expressed and the mutated, maternal allele will be inactive due to the imprinting mechanism.
The exact function of ε-sarcoglycan has not yet been elucidated at the cellular level. It is known to have a transmembrane domain and to be located at the plasma membrane. Other sarcoglycans form the dystrophin-glycoprotein complex that links the cytoskeleton to the extracellular matrix and mutations in these genes cause limb-girdle muscular dystrophies [66]. The sarcoglycan complex has extensively been studied in muscle cells. However, the disease-related role of ε-sarcoglycan has to be related to expression in the brain. In contrast to other sarcoglycans, ε-sarcoglycan is also expressed in astrocytes, endothelial cells, and pericytes. It may play an important role at the blood-brain barrier (BBB) [67•]. Of note, dysfunction of the BBB has also been implicated in primary familial brain calcification (PFBC) due to mutations in PDGFB [68••] and its receptor PDGFRB [69], where dystonia is part of the phenotypic spectrum [70].
Confirmatory Evidence for a Role of “Older” Genes in Dystonia
Several genes for dystonia seem to be extremely rare and could not be unequivocally confirmed for many years. This includes for instance the PRKRA [71] or TUBB4A genes [72, 73]. Rare variants in other genes such as ANO3 [74] have not only been identified in a number of patients but also frequently in controls [75]. A third scenario is clear linkage finding but lack of knowledge of the exact underlying genetic cause, as for X-linked dystonia-parkinsonism (XDP [76]) despite extensive analyses. In this section, we will briefly illustrate these four examples.
DYT-PRKRA
The first biallelic mutations in the PRKRA gene (protein activator of interferon-induced protein kinase EIF2AK2) were reported in 2008 [71] in patients with a dystonia-parkinsonism syndrome. No convincing biallelic mutations were published for the next 7 years. However, recently, a number of reports described single families with mainly homozygous mutations in PRKRA [77•] (Table 1). Most of the reported patients carry the same missense mutation (c.665C > T, p.Pro222Leu, rs121434410) that seems to result from a shared founder [77•]. The phenotype includes early-onset generalized dystonia, often with laryngeal dystonia; tongue protrusion, prominent oro-mandibular involvement, dysphagia, and retrocollis. Parkinsonian features are mild (or even absent) and do not respond to levodopa therapy [77•]. PRKRA was reported as a stress-response gene and encodes a protein kinase with largely unknown function [71].
DYT-TUBB4A
Mutations in the TUBB4A gene (tubulin beta 4A class IVa) have been described in 2013 in a large Australian DYT4 family [72, 73]. The phenotype is rather unique including severe generalized dystonia with spasmodic dysphonia and craniocervical dystonia as well as extrusional tongue dystonia and a “hobby horse gait” in some patients [78]. Thus, it is not surprising that TUBB4A mutations are an extremely rare cause of dystonia [79] and only one novel 3-bp in-frame deletion was recently found in a patient with cervical dystonia (unpublished data). Similarly, as for ATP1A3 mutations, there is a broad clinical spectrum linked to TUBB4A mutations ranging from isolated dystonia to hypomyelination and brain atrophy with the latter being by far the most common disease manifestation. Whether this represents pleiotropy or a continuous phenotypic spectrum is still under debate [80]. Beta-tubulin is an essential component of microtubules that form the cytoskeleton and serve diverse cellular functions.
DYT-ANO3
Mutations in the ANO3 gene (anoctamin 3) have first been reported in 2012 in patients with predominantly craniocervical dystonia with a broad range of ages of onset [74]. Mutations were detected in about 1% of dystonia patients including small families with segregation of ANO3 variants (Table 1). Notably, a large number of missense variants can be found in variant databases and in healthy individuals [75]. However, a pathogenic role of ANO3 mutations has recently been supported by the description of additional families and a de novo mutation in a patient with dystonia and myoclonic jerks [11•]. Indeed, the presence of de novo mutations in a given disease gene usually supports pathogenicity of the variant [81]. ANO3 encodes a transmembrane protein that belongs to a family of calcium-activated chloride channels and thus may play a role in signal transduction, an important pathway involved in dystonia [15].
DYT-TAF1
The exact mutation causing X-linked dystonia-parkinsonism (XDP; DYT3; lubag) is not yet known but several variants in a disease haplotype segregate with the disease. XDP is the only known X-linked form of an isolated or combined dystonia, endemic to an island of the Philippines, shows neurodegeneration, and seems to be fully penetrant [82]. The disease-linked haplotype could recently be narrowed to a region <300 kb [83] containing the TAF1 gene (TATA box-binding protein-associated factor 1). All seven known disease-specific change (DSC) affect intronic or intergenic regions of or surrounding TAF1 but despite genome sequencing, no additional rare, protein-changing variant in the linked region was found among XDP patients [83]. This supports the hypothesis that changes in the expression of certain isoforms of TAF1 may be causative for XDP [84]. This is further underlined by the detection of missense mutations in TAF1 in patients with a severe neurodevelopmental disorder including dystonic features [85••]. TAF1 encodes one subunit of the transcription factor complex IID. Of note, TATA box-binding protein (TBP), which is encoded by the TBP gene that is mutated in spinocerebellar ataxia type 17 (SCA17), is also part of this transcription factor complex, and dystonia is part of the phenotypic spectrum of SCA17 [86].
Novel Genes for Monogenic Dystonia
Due to the advent of next-generation sequencing, new dystonia genes are currently being reported almost monthly. Many of these genes are responsible for complex forms of dystonia, often accompanied by developmental delay and seizures. Because of space constraints, the authors can only illustrate here a few selected examples not necessarily representing the full spectrum of these novel genes implicated in dystonia.
Mutations in ADCY5 in Patients with Choreatic and Dystonic Features (CHOR/DYT-ADCY5)
Although initially described in 2012 in a single family with dyskinesia and facial myokymia [87], mutations in the ADCY5 gene (adenylate cyclase 5) garnered increasing attention of movement disorder specialists and researchers when it was found in a family with chorea and dystonia in 2015 [88]. Since then, more than 20 mutation carriers have been reported often with de novo mutations and a wide range of clinical manifestations including childhood-onset paroxysmal or persistent choreatic and/or dystonic features, and developmental delay in some cases [50•, 89]. ADCY5 encodes an enzyme that is responsible for the synthesis of cAMP, plays a role in signaling, and is functionally linked to Gαolf. Mutations in ADCY5 cluster within regions encoding the C1 or C2 domain of the protein.
Mutations in GNB1 in patients with Developmental Delay, Hypotonia, and Dystonia
Several de novo mutations in the GNB1 gene (G protein subunit beta 1), encoding a subunit of a guanine nucleotide-binding proteins, have been reported in patients with neurodevelopmental disability and a wide range of additional symptoms and signs including hypotonia, seizures, and dystonia [90••]. This gene has been independently confirmed and another de novo mutation was reported in a patient with prominent dystonic postures [91]. Notably, almost all mutations in GNB1 are located in exons 6 and 7 and thus cluster at the binding surface for interactions with Gα and various downstream effectors. Guanine nucleotide-binding protein form heterotrimeric complexes consisting of the subunits α, β, and γ and play a role in signaling such as GNAL and ADCY5.
Mutations in GNAO1 in Patients with Developmental Delay and Dystonia
More than 20 patients with mostly de novo mutations in the GNAO1 gene (G protein subunit alpha o1) have been reported in the literature [92]. As for other genes of complex dystonia, mutations in GNAO1 are also associated with a wide range of phenotypes including epileptic encephalopathy [93], developmental delay without seizures, and chorea as well as dystonia in a subset of patients. There seems to be a genotype-phenotype correlation with mutations at positions Arg209 and Glu246 leading to the milder phenotype without seizures [92]. Like GNAL and GNB1, GNAO1 also encodes a subunit of a G protein and thus functions in signaling.
Mutations in VAC14 in Patients with Sudden Onset of Developmental Regression and Dystonia
Biallelic mutations in the VAC14 gene (Vac14, PIKFYVE complex component) comprising of four different mutations have recently been reported in two unrelated children from two different families. The disease had a sudden onset of a progressive neurological disorder and regression of developmental milestones at the age of 1–3 years. Both children developed a movement disorder with dystonia, became nonambulatory and nonverbal, and exhibited striatal abnormalities on MRI [94]. VAC14 encodes a subunit of a trimolecular complex (together with PIKFYVE and FIG4) that regulates the level of PI(3,5)P2 which is a signaling lipid localized to the cytoplasmic surface of endolysosomal vesicles. Of note, mutations of FIG4 lead to neurological disorders such as Charcot-Marie-Tooth type 4J neuropathy [94]. Although we were also able to identify a compound-heterozygous missense mutations in a patient with dystonia, anarthria, tetraspasticity, and supranuclear palsy (unpublished data), the role of VAC14 mutations in dystonia awaits further confirmation.
Mutations in TBCD in Patients with Developmental Delay, Seizures, Microcephaly, and Dystonia
Two homozygous missense mutations in the TBCD gene (tubulin-folding cofactor D) have recently been described in four patients from two small consanguineous families [95]. The phenotype of an infantile neurodegenerative disorder was characterized by loss of developmental milestones at the age of 1–2 years, seizures, acquired microcephaly, and dystonia [95]. TBCD seems to be important for cortical cell proliferation and radial migration in the developing mouse brain [95]. Interestingly, TBCD encodes a tubulin-specific chaperon that is required for the de novo assembly of the α-/β-tubulin heterodimer. Of note, the dystonia gene TUBB4A encodes a β-tubulin. Independent replication of TBCD mutations in (complex) dystonia patients is warranted.
Loss-of-Function Mutations in KMT2B in Patients with Dystonia
Very recently, two groups independently identified mainly truncating but also missense mutations in KMT2B as a cause of early-onset generalized dystonia often accompanied by other symptoms including intellectual disability, microcephaly, psychiatric features, dysmorphia, or skin lesions [96••, 97••]. The majority of the mutations occurred de novo, but rarely autosomal dominant inheritance with variable expressivity was also observed. KMT2B mutations may account for up to 10% of early-onset generalized dystonia [96••] but further validation is warranted. KMT2B is a variable gene and variants that only mildly impact on the functionality of the encoded protein, such as missense changes, may not be linked to dystonia. Of note, patients with copy number variations on chromosome 19q13 including the KMT2B gene also developed dystonic features [96••, 97••]. KMT2B encodes the lysine-specific histone methyltransferase 2B and thus links disordered histone modification and chromatin states to the disease mechanism of dystonia.
Challenges for Gene Identification in the Era of Next-Generation Sequencing
In the past two decades, a number of dystonia genes have been reported with an exponential increase during the past 5 years due to the introduction of NGS techniques. With this high-throughput approach, the challenge is not any longer to identify a pathogenic mutation at all but to select the pathogenic variant from thousands of candidates [98, 99]. There are a number of potential pitfalls associated with NGS, which may easily lead to false-positive and false-negative results. Accordingly, not all of the reported dystonia genes hold up and, in some cases, there is even convincing evidence of false-positive reports [100]. There are hints that can help to distinguish true-positive from false-positive new disease genes with high likelihood but not with certainty. These hints include (I) independent confirmation (which may lag behind the initial description of the gene for several years as in the case of PRKRA), (II) functional studies (which may be difficult if the function of the protein is not yet clear such as for TOR1A), and (III) variability of the gene. For the latter, absolute numbers can help since it is per se more likely to find more (rare) variants in a large gene (Fig. 1).

Current status of replication/confirmation of reported isolated and combined dystonia genes. The figure illustrates the growing number of known dystonia genes in chronological order. While the upper five genes were all identified before the era of NGS, have been confirmed unequivocally in replication studies, and functional changes of mutant proteins have been demonstrated, this picture changed after the introduction of NGS to the field as indicated by the red arrow. Several of the novel dystonia genes have not yet been confirmed in independent studies. For CIZ1, CACNA1B, COL6A3, and HPCA, subsequent screening studies did not identify any additional causative mutations (as indicated by the cross). A possible indication with respect to credibility of novel genes may be reflected by the number of rare variants within a given gene. The numbers in the fourth column are taken from the database of the Exome Aggregation Consortium (ExAC at http://exac.broadinstitute.org/) and are based on more than 60,000 individuals. Of note, while the confirmed genes (as indicated by a check mark) bear an average of 112 rare variants, this number increases for the yet unconfirmed genes (indicated by a question mark) to 574 variants (adapted from [15]), suggesting that additional follow-up investigations are warranted. Check marks indicate “confirmed” while crosses indicate lack of replication or confirmatory functional studies
Genetic Risk Factors for Dystonia
Although genetic risk factors probably play a major role in dystonia, their elucidation is still limited. A number of common genetic variants (polymorphisms) have been investigated as genetic risk factors in candidate gene association studies. This included variants within genes from the dopamine pathway such as dopamine transporter and receptor genes, brain-derived neurotrophic factor (BDNF), and genes involved in monogenic forms of dystonia; however, the results of these studies were overall inconclusive [101]. An alternative way to identify genetic risk variants in a hypothesis-free approach is large-scale genome-wide association studies (GWAS). Surprisingly, only two small GWAS have been reported for dystonia so far. In one study, variants in the sodium leak channel (NALCN) were found as a risk factor in cervical dystonia [102]; in another study, a variant in arylsulfatase G (ARSG) gene was reported to be more frequent among patients with musician´s dystonia [103]. However, replication of these results and additional studies are clearly warranted.
Perspectives
The identification of new pathogenic variants expanded our understanding of disease mechanisms and the pathophysiological basis of dystonia [15] and also impacts on the diagnostic yield of genetic testing. The more genes are known, the more patients can receive a genetic diagnosis and information about the origin of their disease. This is of high clinical importance, as uncertainty is sometimes even more difficult to cope with than an established diagnosis even of a disease with an unfavorable outcome. With the increasing number of genetic causes of dystonia, it is also becoming more and more efficient to use exome sequencing for diagnostic work-up rather than single gene analyses and multiple MRI scans and other often non-specific ancillary tests [98]. Especially in childhood-onset diseases, exome sequencing of parent-child trios have proven a powerful tool [104]. Not only do such trio analyses allow for the detection of de novo mutations but they also enable easy detection of biallelic, including compound-heterozygous, mutations.
In recent years, much emphasis has been placed on the identification of new genes, however, the functional characterization of these genes to understand the disease mechanisms underlying dystonia lags behind, calling for concerted efforts in this regard.
Conclusions
More than 20 years of research in dystonia genetics have led to the identification of a number of monogenic causes of dystonia and thereby provided important insights into its pathophysiology. There are a rapidly increasing number of novel dystonia genes for which careful validation and functional studies are warranted. The expanded knowledge of genetic causes of dystonia improves genetic testing and enables genetic counseling and gene-specific treatment in some cases.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Oppenheim H. Ueber eigenenartige Krampfkrankheit des kindlichen und jugendlichen Alters (Dysbasia lordotica progressiva, Dystonia Musculorum Deformans). Neurol Centrabl. 1911;30:1090.
• C. Klein, S. Fahn, Translation of Oppenheim’s 1911 paper on dystonia, Mov Disord 28 (2013) 851-862. This is a translation and commentary of Oppenheim’s landmark paper coining the term dystonia and most likely describing the first cases of generalized dystonia due to mutations in Tor1A (formerly DYT1) and suspecting a genetic etiology.
Steeves TD, Day L, Dykeman J, Jette N, Pringsheim T. The prevalence of primary dystonia: a systematic review and meta-analysis. Mov Disord. 2012;27:1789–96.
• Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013;28:863–73. This is an important paper that revises the definition and classification of dystonia based on a consensus outcome of an international expert panel. The new classification contains two axes: clinical characteristics and etiology. The latter one replaces the terms “primary” and “dystonia-plus” dystonia with “isolated” and “combined” dystonia, respectively.
Wang L, Chen Y, Hu B, Hu X. Late-onset primary dystonia in Zhejiang province of China: a service-based epidemiological study. Neurol Sci. 2016;37:111–6.
L. Williams, E. McGovern, O. Kimmich, A. Molloy, I. Beiser, J.S. Butler, F. Molloy, P. Logan, D.G. Healy, T. Lynch, R. Walsh, L. Cassidy, P. Moriarty, H. Moore, T. McSwiney, C. Walsh, S. O’Riordan, M. Hutchinson, Epidemiological, clinical and genetic aspects of adult onset isolated focal dystonia in Ireland, Eur J Neurol (2016).
Molloy A, Williams L, Kimmich O, Butler JS, Beiser I, McGovern E, et al. Sun exposure is an environmental factor for the development of blepharospasm. J Neurol Neurosurg Psychiatry. 2016;87:420–4.
Schmidt A, Jabusch HC, Altenmuller E, Hagenah J, Bruggemann N, Lohmann K, et al. Etiology of musician’s dystonia: familial or environmental? Neurology. 2009;72:1248–54.
Groen JL, Kallen MC, van de Warrenburg BP, Speelman JD, van Hilten JJ, Aramideh M, et al. Phenotypes and genetic architecture of focal primary torsion dystonia. J Neurol Neurosurg Psychiatry. 2012;83:1006–11.
van Egmond ME, Kuiper A, Eggink H, Sinke RJ, Brouwer OF, Verschuuren-Bemelmans CC, et al. Dystonia in children and adolescents: a systematic review and a new diagnostic algorithm. J Neurol Neurosurg Psychiatry. 2015;86:774–81.
• M. Zech, S. Boesch, A. Jochim, S. Weber, T. Meindl, B. Schormair, T. Wieland, C. Lunetta, V. Sansone, M. Messner, J. Mueller, A. Ceballos-Baumann, T.M. Strom, R. Colombo, W. Poewe, B. Haslinger, J. Winkelmann, Clinical exome sequencing in early-onset generalized dystonia and large-scale resequencing follow-up, Mov Disord (2016). This is a relevant paper in two respects: first, it uses exome sequencing to detect the cause of dystonia in a small group of patients underlining the heterogeneity of dystonia. Second, it described the first de novo mutation in ANO3 providing increasing evidence for a pathogenic role of mutations in this gene.
•• Marras C, Lang A, van de Warrenburg BP, Sue CM, Tabrizi SJ, Bertram L, et al. Nomenclature of genetic movement disorders: recommendations of the international Parkinson and movement disorder society task force. Mov Disord. 2016;31:436–57. This paper is of major importance since it suggests a new nomenclature system for genetic movement disorders based on extensive considerations and discussions among an expert panel and members of the Movement Disorder Society.
Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–8.
Marras C, Lohmann K, Lang A, Klein C. Fixing the broken system of genetic locus symbols: Parkinson disease and dystonia as examples. Neurology. 2012;78:1016–24.
Domingo A, Erro R, Lohmann K. Novel dystonia genes: clues on disease mechanisms and the complexities of high-throughput sequencing. Mov Disord. 2016;31:471–7.
Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41:286–8.
Fuchs T, Saunders-Pullman R, Masuho I, Luciano MS, Raymond D, Factor S, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet. 2013;45:88–92.
Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8:236–42.
de Carvalho Aguiar P, Sweadner KJ, Penniston JT, Zaremba J, Liu L, Caton M, et al. Mutations in the Na+/K+-ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 2004;43:169–75.
Zimprich A, Grabowski M, Asmus F, Naumann M, Berg D, Bertram M, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet. 2001;29:66–9.
LeDoux MS. The genetics of dystonia. Adv Genet. 2012;79:35–85.
Bressman SB, Sabatti C, Raymond D, de Leon D, Klein C, Kramer PL, et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology. 2000;54:1746–52.
Dobricic V, Kresojevic N, Zarkovic M, Tomic A, Marjanovic A, Westenberger A, et al. Phenotype of non-c.907_909delGAG mutations in TOR1A: DYT1 dystonia revisited. Parkinsonism Relat Disord. 2015;21:1256–9.
Vulinovic F, Lohmann K, Rakovic A, Capetian P, Alvarez-Fischer D, Schmidt A, et al. Unraveling cellular phenotypes of novel TorsinA/TOR1A mutations. Hum Mutat. 2014;35:1114–22.
Zech M, Jochim A, Boesch S, Weber S, Meindl T, Peters A, et al. Systematic TOR1A non-c.907_909delGAG variant analysis in isolated dystonia and controls. Parkinsonism Relat Disord. 2016;31:119–23.
Hettich J, Ryan SD, de Souza ON, Saraiva Macedo Timmers LF, Tsai S, Atai NA, et al. Biochemical and cellular analysis of human variants of the DYT1 dystonia protein, TorsinA/TOR1A. Hum Mutat. 2014;35:1101–13.
Kabakci K, Hedrich K, Leung JC, Mitterer M, Vieregge P, Lencer R, et al. Mutations in DYT1: extension of the phenotypic and mutational spectrum. Neurology. 2004;62:395–400.
• Brüggemann N, Kühn A, Schneider SA, Kamm C, Wolters A, Krause P, et al. Short- and long-term outcome of chronic pallidal neurostimulation in monogenic isolated dystonia. Neurology. 2015;84:895–903. This paper provides a translational aspect on the partially differential clinical outcome after deep brain stimulation in carriers of mutations in different dystonia genes.
Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA. 2004;101:847–52.
B.A. Sosa, F.E. Demircioglu, J.Z. Chen, J. Ingram, H.L. Ploegh, T.U. Schwartz, How lamina-associated polypeptide 1 (LAP1) activates Torsin, Elife 3 (2014) e03239.
Naismith TV, Dalal S, Hanson PI. Interaction of torsinA with its major binding partners is impaired by the dystonia-associated DeltaGAG deletion. J Biol Chem. 2009;284:27866–74.
F.E. Demircioglu, B.A. Sosa, J. Ingram, H.L. Ploegh, T.U. Schwartz, Structures of TorsinA and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia, Elife 5 (2016).
•• Grillet M, Dominguez Gonzalez B, Sicart A, Pottler M, Cascalho A, Billion K, et al. Torsins are essential regulators of cellular lipid metabolism. Dev Cell. 2016;38:235–47. This is a very important study that links Torsin mutations to dysregulated cellular lipid metabolism and thus suggests a novel disease mechanism in dystonia.
• V.G. Shakkottai, A. Batla, K. Bhatia, W.T. Dauer, C. Dresel, M. Niethammer, D. Eidelberg, R.S. Raike, Y. Smith, H.A. Jinnah, E.J. Hess, S. Meunier, M. Hallett, R. Fremont, K. Khodakhah, M.S. LeDoux, T. Popa, C. Gallea, S. Lehericy, A.C. Bostan, P.L. Strick, Current opinions and areas of consensus on the role of the cerebellum in dystonia, Cerebellum (2016). This is an important review summarizing and discussing data on the neuroanatomical site of origin of dystonia. It illustrates that the cerebellum plays a role in the pathophysiology of dystonia, but it is probably neither the primary nor sole relevant neuroanatomical structure.
E. Premi, M. Diano, S. Gazzina, F. Cauda, V. Gualeni, M. Tinazzi, M. Fiorio, P. Liberini, C. Lazzarini, S. Archetti, G. Biasotto, M. Turla, V. Bertasi, M. Cotelli, R. Gasparotti, A. Padovani, B. Borroni, Functional connectivity networks in asymptomatic and symptomatic DYT1 carriers, Mov Disord (2016).
Vanni V, Puglisi F, Bonsi P, Ponterio G, Maltese M, Pisani A, et al. Cerebellar synaptogenesis is compromised in mouse models of DYT1 dystonia. Exp Neurol. 2015;271:457–67.
Blanchard A, Ea V, Roubertie A, Martin M, Coquart C, Claustres M, et al. DYT6 dystonia: review of the literature and creation of the UMD Locus-Specific Database (LSDB) for mutations in the THAP1 gene. Hum Mutat. 2011;32:1213–24.
Lohmann K, Uflacker N, Erogullari A, Lohnau T, Winkler S, Dendorfer A, et al. Identification and functional analysis of novel THAP1 mutations. Eur J Hum Genet. 2012;20:171–5.
Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saunders-Pullman R. Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol. 2009;8:441–6.
Paudel R, Li A, Hardy J, Bhatia KP, Houlden H, Holton J. DYT6 dystonia: a neuropathological study. Neurodegener Dis. 2016;16:273–8.
Krause P, Bruggemann N, Volzmann S, Horn A, Kupsch A, Schneider GH, et al. Long-term effect on dystonia after pallidal deep brain stimulation (DBS) in three members of a family with a THAP1 mutation. J Neurol. 2015;262:2739–44.
Ortiz-Virumbrales M, Ruiz M, Hone E, Dolios G, Wang R, Morant A, et al. Dystonia type 6 gene product Thap1: identification of a 50 kDa DNA-binding species in neuronal nuclear fractions. Acta Neuropathol Commun. 2014;2:139.
Gavarini S, Cayrol C, Fuchs T, Lyons N, Ehrlich ME, Girard JP, et al. Direct interaction between causative genes of DYT1 and DYT6 primary dystonia. Ann Neurol. 2010;68:549–53.
Kaiser FJ, Osmanoric A, Rakovic A, Erogullari A, Uflacker N, Braunholz D, et al. The dystonia gene DYT1 is repressed by the transcription factor THAP1. Ann Neurol. 2010;68:554–9. DYT6.
LeDoux MS, Vemula SR, Xiao J, Thompson MM, Perlmutter JS, Wright LJ, et al. Clinical and genetic features of cervical dystonia in a large multicenter cohort. Neurol Genet. 2016;2:e69.
Vemula SR, Puschmann A, Xiao J, Zhao Y, Rudzinska M, Frei KP, et al. Role of Galpha(olf) in familial and sporadic adult-onset primary dystonia. Hum Mol Genet. 2013;22:2510–9.
• Masuho I, Fang M, Geng C, Zhang J, Jiang H, Ozgul RK, et al. Homozygous GNAL mutation associated with familial childhood-onset generalized dystonia. Neurol Genet. 2016;2:e78. This is an interesting case report on a family with homozygous mutations in an actually dominantly inherited disorder. The data are not only descriptive on the phenotypic level but are accompanied by detailed functional analysis of the mutation underlining the importance of functional studies.
Opladen T, Hoffmann G, Horster F, Hinz AB, Neidhardt K, Klein C, et al. Clinical and biochemical characterization of patients with early infantile onset of autosomal recessive GTP cyclohydrolase I deficiency without hyperphenylalaninemia. Mov Disord. 2011;26:157–61.
Kumar KR, Lohmann K, Masuho I, Miyamoto R, Ferbert A, Lohnau T, et al. Mutations in GNAL: a novel cause of craniocervical dystonia. JAMA Neurol. 2014;71:490–4.
• Chen DH, Meneret A, Friedman JR, Korvatska O, Gad A, Bonkowski ES, et al. ADCY5-related dyskinesia: broader spectrum and genotype-phenotype correlations. Neurology. 2015;85:2026–35. This study reports many cases of ADCY5-related dyskinesia with a wide range of hyperkinetic abnormal movements.
V. Tadic, M. Kasten, N. Bruggemann, S. Stiller, J. Hagenah, C. Klein, Dopa-responsive dystonia revisited: diagnostic delay, residual signs, and nonmotor signs, Arch Neurol (2012) 1-5.
Lewthwaite AJ, Lambert TD, Rolfe EB, Olgiati S, Quadri M, Simons EJ, et al. Novel GCH1 variant in dopa-responsive dystonia and Parkinson’s disease. Parkinsonism Relat Disord. 2015;21:394–7.
Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, Polke JM, et al. Parkinson’s disease in GTP cyclohydrolase 1 mutation carriers. Brain. 2014;137:2480–92.
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46:989–93.
Hagenah J, Saunders-Pullman R, Hedrich K, Kabakci K, Habermann K, Wiegers K, et al. High mutation rate in dopa-responsive dystonia: detection with comprehensive GCHI screening. Neurology. 2005;64:908–11.
Douglas G, Hale AB, Crabtree MJ, Ryan BJ, Hansler A, Watschinger K, et al. A requirement for Gch1 and tetrahydrobiopterin in embryonic development. Dev Biol. 2015;399:129–38.
Brüggemann N, Spiegler J, Hellenbroich Y, Opladen T, Schneider SA, Stephani U, et al. Beneficial prenatal levodopa therapy in autosomal recessive guanosine triphosphate cyclohydrolase 1 deficiency. Arch Neurol. 2012;69:1071–5.
Brashear A, Dobyns WB, de Carvalho Aguiar P, Borg M, Frijns CJ, Gollamudi S, et al. The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain. 2007;130:828–35.
• Sweney MT, Newcomb TM, Swoboda KJ. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, alternating hemiplegia of childhood, rapid-onset dystonia-parkinsonism, CAPOS and beyond. Pediatr Neurol. 2015;52:56–64. This important article illustrates the broad phenotypic spectrum of mutations in a single gene, i.e., ATP1A3.
Holm TH, Isaksen TJ, Glerup S, Heuck A, Bottger P, Fuchtbauer EM, et al. Cognitive deficits caused by a disease-mutation in the alpha3 Na(+)/K(+)-ATPase isoform. Sci Rep. 2016;6:31972.
M. Hully, J. Ropars, L. Hubert, N. Boddaert, M. Rio, M. Bernardelli, I. Desguerre, V. Cormier-Daire, A. Munnich, P. de Lonlay, L. Reilly, C. Besmond, N. Bahi-Buisson, Mosaicism in ATP1A3-related disorders: not just a theoretical risk, Neurogenetics (2016).
Grünewald A, Djarmati A, Lohmann-Hedrich K, Farrell K, Zeller JA, Allert N, et al. Myoclonus-dystonia: significance of large SGCE deletions. Hum Mutat. 2008;29:331–2.
Peall KJ, Kurian MA, Wardle M, Waite AJ, Hedderly T, Lin JP, et al. SGCE and myoclonus dystonia: motor characteristics, diagnostic criteria and clinical predictors of genotype. J Neurol. 2014;261:2296–304.
Peall KJ, Dijk JM, Saunders-Pullman R, Dreissen YE, van Loon I, Cath D, et al. Psychiatric disorders, myoclonus dystonia and SGCE: an international study. Ann Clin Transl Neurol. 2016;3:4–11.
Müller B, Hedrich K, Kock N, Dragasevic N, Svetel M, Garrels J, et al. Evidence that paternal expression of the epsilon-sarcoglycan gene accounts for reduced penetrance in myoclonus-dystonia. Am J Hum Genet. 2002;71:1303–11.
Hack AA, Groh ME, McNally EM. Sarcoglycans in muscular dystrophy. Microsc Res Tech. 2000;48:167–80.
• Boulay AC, Saubamea B, Cisternino S, Mignon V, Mazeraud A, Jourdren L, et al. The sarcoglycan complex is expressed in the cerebrovascular system and is specifically regulated by astroglial Cx30 channels. Front Cell Neurosci. 2015;9:9. This functional study highlights a potential disease mechanism of mutations in the SGCE gene by linking the sarcoglycan complex to the cerebrovascular system.
•• A. Keller, A. Westenberger, M.J. Sobrido, M. Garcia-Murias, A. Domingo, R.L. Sears, R.R. Lemos, A. Ordonez-Ugalde, G. Nicolas, J.E. da Cunha, E.J. Rushing, M. Hugelshofer, M.C. Wurnig, A. Kaech, R. Reimann, K. Lohmann, V. Dobricic, A. Carracedo, I. Petrovic, J.M. Miyasaki, I. Abakumova, M.A. Mae, E. Raschperger, M. Zatz, K. Zschiedrich, J. Klepper, E. Spiteri, J.M. Prieto, I. Navas, M. Preuss, C. Dering, M. Jankovic, M. Paucar, P. Svenningsson, K. Saliminejad, H.R. Khorshid, I. Novakovic, A. Aguzzi, A. Boss, I. Le Ber, G. Defer, D. Hannequin, V.S. Kostic, D. Campion, D.H. Geschwind, G. Coppola, C. Betsholtz, C. Klein, J.R. Oliveira, Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice, Nat Genet (2013). This comprehensive study elucidated a frequent cause of primary familial brain calcification by identification of mutations in the PDGFB gene. It not only reported the cause of the disease in several multiplex families but also demonstrated corresponding brain calcifications in mouse models at different ages and linked this complex form of dystonia to dysfunction of the blood brain barrier.
Nicolas G, Pottier C, Maltete D, Coutant S, Rovelet-Lecrux A, Legallic S, et al. Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology. 2013;80:181–7.
Westenberger A, Klein C. The genetics of primary familial brain calcifications. Curr Neurol Neurosci Rep. 2014;14:490.
Camargos S, Scholz S, Simon-Sanchez J, Paisan-Ruiz C, Lewis P, Hernandez D, et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008;7:207–15.
Hersheson J, Mencacci NE, Davis M, Macdonald N, Trabzuni D, Ryten M, et al. Mutations in the autoregulatory domain of beta-tubulin 4a cause hereditary dystonia. Ann Neurol. 2013;73:546–53.
Lohmann K, Wilcox RA, Winkler S, Ramirez A, Rakovic A, Park JS, et al. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann Neurol. 2013;73:537–45.
Charlesworth G, Plagnol V, Holmstrom KM, Bras J, Sheerin UM, Preza E, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet. 2012;91:1041–50.
Zech M, Gross N, Jochim A, Castrop F, Kaffe M, Dresel C, et al. Rare sequence variants in ANO3 and GNAL in a primary torsion dystonia series and controls. Mov Disord. 2014;29:143–7.
Nolte D, Niemann S, Muller U. Specific sequence changes in multiple transcript system DYT3 are associated with X-linked dystonia parkinsonism. Proc Natl Acad Sci USA. 2003;100:10347–52.
• Quadri M, Olgiati S, Sensi M, Gualandi F, Groppo E, Rispoli V, et al. PRKRA mutation causing early-onset generalized dystonia-parkinsonism (DYT16) in an Italian family. Mov Disord. 2016;31:765–7. This interesting paper confirms biallelic mutations in the PRKRA gene as a cause of dystonia-parkinsonism and demonstrates a founder effect.
Wilcox RA, Winkler S, Lohmann K, Klein C. Whispering dysphonia in an Australian family (DYT4): a clinical and genetic reappraisal. Mov Disord. 2011;26:2404–8.
Zech M, Boesch S, Jochim A, Graf S, Lichtner P, Peters A, et al. Large-scale TUBB4A mutational screening in isolated dystonia and controls. Parkinsonism Relat Disord. 2015;21:1278–81.
Erro R, Hersheson J, Ganos C, Mencacci NE, Stamelou M, Batla A, et al. H-ABC syndrome and DYT4: variable expressivity or pleiotropy of TUBB4 mutations? Mov Disord. 2015;30:828–33.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Lee LV, Pascasio FM, Fuentes FD, Viterbo GH. Torsion dystonia in Panay. Philippines Adv Neurol. 1976;14:137–51.
Domingo A, Westenberger A, Lee LV, Braenne I, Liu T, Vater I, et al. New insights into the genetics of X-linked dystonia-parkinsonism. Eur J Hum Genet. 2015;23:1334–40. XDP, DYT3.
Domingo A, Amar D, Grutz K, Lee LV, Rosales R, Bruggemann N, et al. Evidence of TAF1 dysfunction in peripheral models of X-linked dystonia-parkinsonism. Cell Mol Life Sci. 2016;73:3205–15.
•• J.A. O’Rawe, Y. Wu, M.J. Dorfel, A.F. Rope, P.Y. Au, J.S. Parboosingh, S. Moon, M. Kousi, K. Kosma, C.S. Smith, M. Tzetis, J.L. Schuette, R.B. Hufnagel, C.E. Prada, F. Martinez, C. Orellana, J. Crain, A. Caro-Llopis, S. Oltra, S. Monfort, L.T. Jimenez-Barron, J. Swensen, S. Ellingwood, R. Smith, H. Fang, S. Ospina, S. Stegmann, N. Den Hollander, D. Mittelman, G. Highnam, R. Robison, E. Yang, L. Faivre, A. Roubertie, J.B. Riviere, K.G. Monaghan, K. Wang, E.E. Davis, N. Katsanis, V.M. Kalscheuer, E.H. Wang, K. Metcalfe, T. Kleefstra, A.M. Innes, S. Kitsiou-Tzeli, M. Rosello, C.E. Keegan, G.J. Lyon, TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations, Am J Hum Genet 97 (2015) 922-932. This manuscript describes bona fide mutations in the TAF1 gene, the gene that is also dysregulated in X-linked dystonia-parkinsonism (XDP) due to several disease-specific changes.
Hagenah JM, Zuhlke C, Hellenbroich Y, Heide W, Klein C. Focal dystonia as a presenting sign of spinocerebellar ataxia 17. Mov Disord. 2004;19:217–20.
Chen YZ, Matsushita MM, Robertson P, Rieder M, Girirajan S, Antonacci F, et al. Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol. 2012;69:630–5.
Carapito R, Paul N, Untrau M, Le Gentil M, Ott L, Alsaleh G, et al. A de novo ADCY5 mutation causes early-onset autosomal dominant chorea and dystonia. Mov Disord. 2015;30:423–7.
F.C. Chang, A. Westenberger, R.C. Dale, M. Smith, H.S. Pall, B. Perez-Duenas, P. Grattan-Smith, R.A. Ouvrier, N. Mahant, B.C. Hanna, M. Hunter, J.A. Lawson, C. Max, R. Sachdev, E. Meyer, D. Crimmins, D. Pryor, J.G. Morris, A. Munchau, D. Grozeva, K.J. Carss, L. Raymond, M.A. Kurian, C. Klein, V.S. Fung, Phenotypic insights into ADCY5-associated disease, Mov Disord (2016).
•• S. Petrovski, S. Kury, C.T. Myers, K. Anyane-Yeboa, B. Cogne, M. Bialer, F. Xia, P. Hemati, J. Riviello, M. Mehaffey, T. Besnard, E. Becraft, A. Wadley, A.R. Politi, S. Colombo, X. Zhu, Z. Ren, I. Andrews, T. Dudding-Byth, A.L. Schneider, G. Wallace, A.B. Rosen, S. Schelley, G.M. Enns, P. Corre, J. Dalton, S. Mercier, X. Latypova, S. Schmitt, E. Guzman, C. Moore, L. Bier, E.L. Heinzen, P. Karachunski, N. Shur, T. Grebe, A. Basinger, J.M. Nguyen, S. Bezieau, K. Wierenga, J.A. Bernstein, I.E. Scheffer, J.A. Rosenfeld, H.C. Mefford, B. Isidor, D.B. Goldstein, Germline De Novo Mutations in GNB1 Cause Severe Neurodevelopmental Disability, Hypotonia, and Seizures, Am J Hum Genet 98 (2016) 1001-1010. This nicely performed screening study elucidated mutations in the GNB1 gene as a rare but recurrent cause of a complex form of dystonia and highlights G protein-mediated signaling as one disease mechanism in dystonia and neurodevelopmental disability.
Steinrucke S, Lohmann K, Domingo A, Rolfs A, Baumer T, Spiegler J, et al. Novel GNB1 missense mutation in a patient with generalized dystonia, hypotonia, and intellectual disability. Neurol Genet. 2016;2:e106.
L.A. Menke, M. Engelen, M. Alders, V.J. Odekerken, F. Baas, J.M. Cobben, Recurrent GNAO1 mutations associated with developmental delay and a movement disorder, J Child Neurol (2016).
Nakamura K, Kodera H, Akita T, Shiina M, Kato M, Hoshino H, et al. De Novo mutations in GNAO1, encoding a Galphao subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet. 2013;93:496–505.
Lenk GM, Szymanska K, Debska-Vielhaber G, Rydzanicz M, Walczak A, Bekiesinska-Figatowska M, et al. Biallelic mutations of VAC14 in pediatric-onset neurological disease. Am J Hum Genet. 2016;99:188–94.
S. Edvardson, G. Tian, H. Cullen, H. Vanyai, L. Ngo, S. Bhat, A. Aran, M. Daana, N. Da’amseh, B. Abu-Libdeh, N.J. Cowan, J. Heng, O. Elpeleg, Infantile neurodegenerative disorder associated with mutations in TBCD, an essential gene in the tubulin heterodimer assembly pathway, Hum Mol Genet (2016).
•• Zech M, Boesch S, Maier EM, Borggraefe I, Vill K, Laccone F, et al. Haploinsufficiency of KMT2B, encoding the lysine-specific histone methyltransferase 2B, results in early-onset generalized dystonia. Am J Hum Genet. 2016;99:1377–87. This paper represents one of the two studies elucidating KMT2B mutations as a relatively common cause of early-onset dystonia.
•• E. Meyer, K.J. Carss, J. Rankin, J.M. Nichols, D. Grozeva, A.P. Joseph, N.E. Mencacci, A. Papandreou, J. Ng, S. Barral, A. Ngoh, H. Ben-Pazi, M.A. Willemsen, D. Arkadir, A. Barnicoat, H. Bergman, S. Bhate, A. Boys, N. Darin, N. Foulds, N. Gutowski, A. Hills, H. Houlden, J.A. Hurst, Z. Israel, M. Kaminska, P. Limousin, D. Lumsden, S. McKee, S. Misra, S.S. Mohammed, V. Nakou, J. Nicolai, M. Nilsson, H. Pall, K.J. Peall, G.B. Peters, P. Prabhakar, M.S. Reuter, P. Rump, R. Segel, M. Sinnema, M. Smith, P. Turnpenny, S.M. White, D. Wieczorek, S. Wiethoff, B.T. Wilson, G. Winter, C. Wragg, S. Pope, S.J. Heales, D. Morrogh, A. Pittman, L.J. Carr, B. Perez-Duenas, J.P. Lin, A. Reis, W.A. Gahl, C. Toro, K.P. Bhatia, N.W. Wood, E.J. Kamsteeg, W.K. Chong, P. Gissen, M. Topf, R.C. Dale, J.R. Chubb, F.L. Raymond, M.A. Kurian, Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia, Nat Genet (2016). This paper represents one of the two studies elucidating KMT2B mutations as a relatively common cause of early-onset dystonia.
K. Lohmann, C. Klein, Next generation sequencing and the future of genetic diagnosis, Neurotherapeutics (2014).
Olgiati S, Quadri M, Bonifati V. Genetics of movement disorders in the next-generation sequencing era. Mov Disord. 2016;31:458–70.
Mencacci NE, R’Bibo L, Bandres-Ciga S, Carecchio M, Zorzi G, Nardocci N, et al. The CACNA1B R1389H variant is not associated with myoclonus-dystonia in a large European multicentric cohort. Hum Mol Genet. 2015;24:5326–9.
Lohmann K, Klein C. Genetics of dystonia: what’s known? what’s new? what’s next? Mov Disord. 2013;28:899–905.
Mok KY, Schneider SA, Trabzuni D, Stamelou M, Edwards M, Kasperaviciute D, et al. Genomewide association study in cervical dystonia demonstrates possible association with sodium leak channel. Mov Disord. 2014;29:245–51.
Lohmann K, Schmidt A, Schillert A, Winkler S, Albanese A, Baas F, et al. Genome-wide association study in musician’s dystonia: a risk variant at the arylsulfatase G locus? Mov Disord. 2014;29:921–7.
Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–75.
Acknowledgments
Katja Lohmann is supported by the DFG (LO 1555/4-1 and LO 1555/3-2). Christine Klein is a recipient of a career development award from the Hermann and Lilly Schilling Foundation. Further, Katja Lohmann and Christine Klein are supported by the German Ministry of Education and Research (BMBF, DYSTRACT consortium, 01GM1514B) and a Research Unit of the DFG (FOR2488).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Katja Lohmann declares no conflict of interest.
Christine Klein has received personal fees from Centogene and from the Else Kroener Fresenius Foundation.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Genetics
Rights and permissions
About this article

Cite this article
Lohmann, K., Klein, C. Update on the Genetics of Dystonia. Curr Neurol Neurosci Rep 17, 26 (2017). https://doi.org/10.1007/s11910-017-0735-0
Published:
DOI: https://doi.org/10.1007/s11910-017-0735-0