
Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is a clinically recognizable and relatively common muscular dystrophy. It is inherited mostly as an autosomal dominant disease or in a minority of cases, in a digenic pattern. The disease manifestation is variable and most likely dependent on genetic and epigenetic factors. We review the history, epidemiology, clinical presentation, and genetics of the disease, present the recently elucidated molecular pathogenesis, discuss the pathology and the possible consequence of the inflammation seen in the muscle biopsies, and consider future treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Facioscapulohumeral dystrophy (FSHD) is the third most common muscular dystrophy after Duchenne muscular dystrophy and myotonic dystrophy. FSHD is a unique and complex genetic disease inherited mostly as an autosomal dominant disease or in a minority of cases, in a digenic pattern. FSHD can manifest as a patchy and slowly progressive disease primarily of skeletal muscle. Although it generally does not affect mortality, it can cause significant disability in up to 20 % of patients over the age of 50—causing lost of independent ambulation and requiring full-time wheelchair use.
History
The initial cases of FSHD were described around the mid-1800s in France. In 1862, Duchenne published a photograph of a typical case [1]. In 1868, he described a “progressive fatty muscular atrophy of childhood beginning around the age of five-to-seven years affecting the face or causing atrophy of muscles, mainly the orbicularis, lips, and cheekbones. After a stationary period of several (2–3) years, it invades the limbs and trunk; or it works in the same way in the adult—that is to say—it follows a downward march, by first attacking the muscles of the upper limbs and trunk and then extends to the lower limbs in a fairly advanced stage” [2].
In 1885, Landouzy and Dejerine published the first case history of a patient that fit Duchenne’s description, with facial weakness at 3, shoulder girdle and upper arm weakness at 17, and subsequent truncal and pelvic girdle weakness before the age of 24 [3]. They aptly named their subsequent eponymous disease: “facioscapulohumeral type of progressive myopathy.” Their case histories of the patient’s relatives (1885–1886) establish an autosomal dominant inheritance pattern with variable penetrance and a pleomorphic presentation. Many family members had mild disease, with one presenting with facial weakness at 9 and symptomatic limb weakness not until 60; and another presenting with shoulder girdle weakness first and facial weakness later.
Further descriptions of the disease in a large family in Utah confirmed the autosomal dominant inheritance pattern [4]. In 1990, the gene locus responsible for FSHD was mapped to 4q35 [5]. In 1992, contraction in the number of macrosatellite repeats at 4q35 was identified as the genetic defect [5, 6]. In 2010, the DUX4 gene, a copy of which is located in each macrosatellite repeat, was found to be aberrantly expressed [7, 8]. This was made possible with the advancement of polymerase chain reaction technology to reliably detect low copies of messenger RNA (mRNA). In 2012 and 2016, mutations in two other genes on different chromosomes resulting in decreased heterochromatin condensation and subsequent DUX4 de-repression were described in small percentage of FSHD families with a normal D4Z4 repeat size [9, 10].
Clinical Presentation
Duchenne’s description in 1868 still fits the classic presentation of the disease. Symptoms are usually noted by parents or become obvious when an affected sibling is diagnosed. Facial weakness, such as children sleeping with their eyes slightly open, is usually the first clinical sign that parents notice. More subtly, facial weakness such as difficulty whistling or sucking on straws is the first clinical sign that patients recognize in retrospect. Winged scapula is usually first noted by parents in late childhood or adolescence. Shoulder girdle weakness with difficulty lifting objects above shoulders is insidious but is the most common symptom that prompts medical attention. Truncal and lower extremity weakness (specifically, weakness of foot dorsiflexion) may be next—resulting in the classic scapuloperoneal distribution of weakness.
The muscular atrophy can result in a characteristic appearance: full or slightly everted lips; winged scapula (most appreciated when arms raised to the shoulder level or pressed against the wall with the hands at shoulder level and elbows straight); straight clavicles, vertical or reversed anterior axillary fold (normally diagonal from the chest to the head of the humerus formed by the pectorals); pectus excavatum (a caved-in chest) [13]; or the Beevor sign resulting from selective weakness of the lower rectus abdominus more so than the upper part.
However, the classic presentation of facial or scapulohumeral weakness occurs in only ∼70–85 %. Facial weakness is spared in 6–18 %. FSHD should be suspected in patchy and asymmetric myopathy as there are many case reports of its manifestation: facial-sparing scapular myopathy, limb-girdle weakness, late-onset distal myopathy after age 50, symmetric brachial weakness, isolated axial weakness, monomelic lower limb atrophy [14].
Bulbar, extraocular, and respiratory muscles tend to be spared. Age of onset is variable with presentations at birth to late life. Penetrance is also variable with 12–30 % of familial cases asymptomatic [4, 15–17]. Prognosis is fair with 20 % of the patients who are wheelchair dependent and ∼1–13 % of the patients requiring noninvasive or invasive respiratory support [18–20].
Genetics
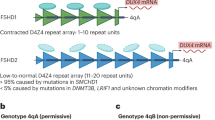
The disease may be the first human inherited neuromuscular disease that results from inappropriate expression of a normally dormant gene, akin to an oncogene. In a normal subtelomere region of chromosome 4q, there are usually 11–100 tandem copies of the 3.3 kilobase (kb) D4Z4 macrosatellite repeat, each containing a single copy of DUX4. DUX4 is normally not expressed in adult somatic tissues including muscle. To cause disease, several factors need to be present: First, at least one D4Z4 repeat must be present. Second, contraction of the D4Z4 repeat array to less than 10 repeat units, which enables chromatin remodeling and decreased methylation. Decreased DNA methylation is associated with a more open chromatin structure—increasing the likelihood that genes in that locus will be expressed. Third and lastly, disease is only manifested in chromosomes carrying a pLAM1 polyadenylation site (A variant) just distal to the last D4Z4 repeat [8]. These factors allow for transcription of the DUX4 gene as well as stabilization of DUX4 mRNA (with the polyadenylated tail allowing for a longer mRNA half-life) and ultimately expression of the transcription factor that is normally repressed (see Fig. 1).

Genetic changes that lead to FSHD. The central component of FSHD molecular pathogenesis is the de-repression of the DUX4 transcription factor, which is present in each D4Z4 repeat (orange triangle). At least one D4Z4 repeat must be present. FSHD is only manifested in chromosomes carrying a permissive haplotype that contains a functional polyadenylation site (green rectangle) just distal to the last D4Z4 repeat. Decreased DNA methylation (red circles) is associated with a more open chromatin structure (open orange triangle). These factors allow for transcription of the DUX4 gene and stabilization of DUX4 mRNA and ultimately expression of the transcription factor that is normally repressed
Whereas 95 % of patient with FSHD have contractions in the D4Z4 repeats (now known as FSHD1), a subpopulation (5 %) of patients, clinically indistinguishable from FSHD1, have a normal number (>10) of D4Z4 repeats. These patients, labeled FSHD2, carry mutations in genes on other chromosomes resulting in hypomethylation of D4Z4 on both copies of 4q35 [21]. As in FSHD1, when such patients carry a permissive haplotype and the associated polyadenylation signal, transcription and expression of DUX4 is enabled and results in disease manifestation (see Fig. 1). Eighty percent of the patients have mutations in the structural maintenance of chromosomal hinge domain 1 (SMCHD1) gene on chromosome 18p [9]. Others carry mutations in DNA methyltransferase 3B (DNMT3B) on chromosome 20q, a gene also associated with immunodeficiency, centromeric instability, and facial anomalies (ICF syndrome) [10]. The inheritance of FSHD2 is digenic as it requires inheritance of two independent genetic events: mutation in a gene regulating chromatin condensation (such as SMCHD1 or DNMT3B) and the presence of permissive polyadenylation signal on 4q35.
Genotype-Phenotype Association
D4Z4 fragment size and methylation status appear to be associated with severity of disease—as shorter D4Z4 fragment sizes result in lower methylation levels and more permissive DUX4 expression. Smaller D4Z4 repeat size is associated with more severe disease [22] as measured by age at diagnosis [23•], age of onset [24], and age at wheelchair dependence [23•]. Genetic modifiers (such as SCHMD1 mutations) that regulate methylation status of D4Z4 repeats also contribute to the severity of the disease [25–27].
Therefore, a classification of disease severity based on genetics has been proposed [28]:
Patients with 1–3 D4Z4 repeats tend to have more severe disease and are more likely to manifest disease. Penetrance is high: ∼85–100 % [15, 17]. Infantile-onset FSHD patients with disease onset before 10 years of age tend to have 1–3 D4Z4 repeats. Facial weakness is not usually severe at onset. These patients can progress to severe diffuse weakness and may not even be able to close their eyes during sleep. They are also at risk of developing extra-muscular manifestations such as hearing loss [29] and vascular retinopathy [30]. Disease manifestation of patients with 1–3 D4Z4 repeats is not restricted to childhood and can also present in adulthood [23•].
Patients with 4–7 D4Z4 repeats tend to have more moderate and variable disease manifestation. Many more patients are asymptomatic gene carriers. Penetrance is lower than the former category: ∼70 % patients with 4–6 D4Z4 repeats were found to be symptomatic by age 60 [17]. Disease onset is later than patients with 1–3 D4Z4 repeats [17]. These patients have a variable course: 60 % with affected legs and 20 % in wheelchairs after 50 years of age [28].
Patients with 7–10 D4Z4 repeats tend to have mild disease as assessed by muscle pathology and strength [31]. None require wheelchair usage. The variation in presentation may be dependent on epigenetic factors such as methylation status affecting DUX4 expression. SMCHD1 mutations can act as genetic modifiers in FSHD1 patients, as evident in mild FSHD1 kindreds with 7–10 D4Z4 repeats who have more severely affected family members due to the presence of a disease-causing SMCHD1 mutation (FSHD1+2) [32•].
Since FSHD2 is the result of mutations in genes that modify chromatin structure, there were concerns that chromatin alterations on chromosomes other than 4q35 may potentially result in additional extra-muscular manifestations. However, FSHD1 and 2 are clinically identical except for the absence, to date, of typical infantile-onset disease with associated hearing and retinal vascular disease in FSHD2 [21]. The severity of disease in FSHD2 may be inversely correlated with the methylation status of the short permissive 4q allele [33].
Molecular Pathogenesis
DUX4 (double homeobox 4) is a transcription factor that appears to be expressed in the spermatagonia in adult male testes and suppressed in adult muscle and other somatic tissue [8]. The forced expression of DUX4 in muscle tissue is highly toxic, leads to apoptosis and oxidative stress, and interferes with normal myogenesis [34–37].
DUX4 expression leads to the induction of a set of genes including PRAMEF1, RFPL2, MBD3L2, TRIM43, KHDC1, and ZSCAN4 [38]. The expression of these genes are undetectable or nearly undetectable in control muscle samples but increased in FSHD muscle samples or DUX4-transfected cell lines [39]. The inappropriate expression of DUX4 protein as well as DUX4-induced proteins, in somatic cells such as the skeletal muscle, may act as antigens triggering the relatively prominent inflammation seen in FSHD muscle.
Muscle Pathology
The histopathology of FSHD muscles is also variable, reflecting the patchy clinical involvement. The histopathology may be normal (at least 10–15 % [40]), shows minimal myopathic changes (see Fig. 2a) or more severe dystrophic changes (see Fig. 2c, d). What is distinctive about FSHD histopathology is the presence of inflammation (see Fig. 2b, d) [41]. Pathologists, given the intensity of the inflammation in some patients, have at times interpreted the histopathology as representing an immune-mediated disease such as polymyositis [42, 43]. The range of inflammation seen in muscle biopsies is variable, as low as 6.3 % of 64 FSHD1 quadriceps biopsies [44] to 46 % [45] or 70 % [41]. The variability may be due to the small sampling size by needle biopsies in certain studies (such as the Statland study).

Variable manifestations of FSHD muscle pathology. Hematoxylin and eosin staining of four different FSHD muscle biopsies, showing: a Minimal changes, variability in fiber size. b More myopathic changes with rounding of fibers and pockets of perivascular inflammation. c Advanced disease with fibro-fatty replacement. d Persistent perivascular inflammation late in the disease in a muscle with advanced pathological changes
The inflammatory cells are also present in a distinct pattern. In Duchenne muscular dystrophy, the inflammatory infiltration is predominantly endomysial—around or in necrotic fibers undergoing phagocytosis. In FSHD, the inflammatory cells are endomysial, surrounding intact fibers, and often perivascular [41, 43, 44, 46, 47]. (also see Fig. 2d).
Muscle MRI studies of FSHD patients suggest that short tau inversion recovery (STIR) hyperintensities may signify a transition state between normal MRI imaging and T1-positive “fatty-replaced” muscle. Two studies also suggest that this STIR hyperintensity represents inflammation [48, 49]. These two studies found that 5/5 biopsies of STIR-positive muscles showed endomysial CD8+ T-cells, perivascular CD4+ T-cells, and CD68+ cells. Moreover, Frisullo et al. found higher percentage of circulating CD8+pSTAT1+, CD8+T-bet+, and CD14+pSTAT1+ cells in the peripheral blood samples of FSHD patients with muscle STIR hyperintensities as compared to FSHD patients without muscle STIR hyperintensity and healthy controls.
Gene expression profiling were performed in four of the five STIR hyperintense muscle biopsies and confirmed in an additional STIR hyperintense muscle [49]. There was a significant upregulation of genes involved in innate immune response (especially, the Toll-like receptor pathway), adaptive immune response, and specifically the classical and alternative complement pathways.
Taken together with the cell culture DUX4 transfection data, one hypothesis may be that DUX4 or one of its target proteins, usually expressed only in immune privileged germline cells, induces an adaptive immune response when these proteins are aberrantly expressed in somatic cells. These findings also raise the question as to whether the inflammation seen in FSHD muscle is a byproduct of the primary pathology or whether it is contributing to the muscle fiber destruction and atrophy.
Diagnosis
Diagnosis is ultimately clinical. Genetic testing is not necessary if family members have genetic confirmation. Genetic testing for FSHD1 looks for contraction of D4Z4 repeats at 4q35. This is done by restriction enzyme digestion of patient genomic DNA and then using a radioactive probe just proximal to the D4Z4 repeats [50]. The resultant fragments resolved by agarose gel electrophoresis reflect the size of the D4Z4 repeats with normal fragment typically >38 kb and fragments containing 1–10 repeats measuring 10–38 kb [51]. If the test is negative, and the clinical suspicion for FSHD is strong, additional testing should be done for FSHD2. This would include first determining the presence of at least one permissive 4qA allele and measuring methylation levels at the D4Z4 repeats followed, if suggestive, by sequencing the SCMHD1 gene (and then the DNMT3B gene). An emerging test for FSHD1 or FSHD2 directly examines the overall methylation status of the D4Z4 repeats [52].
Treatment
Two recently published clinical practice recommendations, one expert opinion based and one evidence-based, provide guidance for the management of individuals with facioscapulohumeral muscular dystrophy [53, 54•]. FSHD currently has no effective treatments. Although many drugs have been tried in clinical trials (prednisone [55, 56], diltiazem [57], albuterol [58, 59], antioxidant supplements with vitamin E, vitamin C, selenomethionine, zinc [60], and a myostatin inhibitor [61]), none showed a clear benefit.
Routine Recommendations
-
Physical/occupational therapy assessment for the need of assistive devices, stretching, range of motion exercises, and safe exercises (of low-resistance/high-repetition exercises, taking into account the patients physical limitations).
-
Low-intensity aerobic exercise is safe and improves function and quality of life [62, 63].
-
Pain assessment with referrals to physical therapy or use of non-steroidal anti-inflammatory drugs for acute pain; referrals to pain clinic, nonpharmacologic therapies (such as exercise and cognitive behavioral therapy), or use of gabapentin or tricyclic antidepressants for chronic pain. This is due to common occurrence of pain and specifically back pain in FSHD patients [64, 65].
Recommended in Pediatric and Adult Patients with 1–3 Repeats
-
Dilated indirect ophthalmoscopy for exudative retinopathy (Coats’ disease).
-
Audiograms in children yet to speak to assess for hearing deficits that may prevent learning and language development.
Recommended in Symptomatic Adults
-
Pulmonary function testing should be obtained at diagnosis, then routinely if abnormal, severely weak proximally, kyphoscoliotic, wheelchair dependent, or with comorbid conditions such as chronic obstructive pulmonary disease or cardiac diseases. If experiencing difficulty breathing, a referral to pulmonary medicine for possible sleep study/overnight oximetry should be initiated.
Scapular Fixation
Scapular fixation by surgery to fix the scapula to the chest wall (typically a combination of wires and bone graft) can increase range of motion in patients with otherwise preserved upper extremity strength. The potential benefit can be assessed pre-surgically by manually pushing and fixing the scapula to the chest wall. The benefit of improved range of motion at the shoulder needs to be weighed against the possible surgical complications: hemo/pneumothorax, pain, infection, and possible decrease in respiratory reserve.
Potential Drug Therapies for FSHD
Drug therapies currently being tested in early phase trials include the newer generation myostatin inhibitor luspatercept and anti-inflammatory biologics (ATYR1940). The ATYR1940 study is based on the hypothesis that immunosuppression of the inflammation seen on muscle pathology may be helpful in slowing disease progression. Small molecules that either block function of DUX4 protein or one of its downstream targets or enhance SMCHD1 function are also being considered.
Gene Therapy for FSHD
The inappropriate expression of DUX4 in FSHD can possibly be suppressed with antisense oligonucleotide (AON) or inhibitory RNA (RNAi) therapies [38, 66]. Suppression of the gene should not be harmful in somatic tissues, as it is not normally expressed. The production of the DUX4 protein is stochastic; therefore, genetic therapies to suppress the gene production would have to be constant. Antisense oligonucleotide therapy is currently under investigation as are methods to deliver AONs as well as RNAi using adeno-associated virus (AAV) vectors. The question will be whether the side effects of gene therapy outweigh the benefits for a non-fatal disease.
Conclusion
FSHD is a common inherited myopathy with a unique and complex molecular pathophysiology. Following the description of the genetic lesion in FSHD, it took 18 years of research to understand how a contraction of a macrosatellite repeat in a heterochromatic region of 4q resulted in FSHD. The elucidation of its molecular pathophysiology and an improved understanding of factors influencing disease progression now allow for clinical trials testing both targeted and non-targeted treatment approaches.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Duchenne GB. Album de photographies pathologiques complementaire de liver intule de l’électrisation localisée. 2nd ed. Paris: JB Baillière et fils; 1862.
Duchenne GB. Recherches sur la paralysie musculaire pseudohypertrophique, ou paralysie myo-sclérosique. Arch Gén Méd. 1868;11:5. 179, 305, 421, 552.
Landouzy L, Dejerine J. De la myopathie atrophique progressive. Rev Med Franc. 1885;5:81–253.
Tyler FH, Stephens FE. Studies in disorders of muscle. II Clinical manifestations and inheritance of facioscapulohumeral dystrophy in a large family. Ann Intern Med. 1950;32:640–60.
Upadhyaya M, Lunt PW, Sarfarazi M, Broadhead W, Daniels J, Owen M, et al. DNA marker applicable to presymptomatic and prenatal diagnosis of facioscapulohumeral disease. Lancet. 1990;336:1320–1.
Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, Dauwerse HG, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26–30.
Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, Nelson AM, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181.
Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camano P, Dauwerse JG, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–3.
Lemmers RJLF, Tawil R, Petek LM, Balog J, Block GJ, Santen GWE, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–4.
van den Boogaard ML, Lemmers RJ, Balog J, Wohlgemuth M, Auranen M, Mitsuhashi S, et al. Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am J Hum Genet. 2016;98(5):1020–9.
Mostacciuolo ML, Pastorello E, Vazza G, Miorin M, Angelini C, Tomelleri G, et al. Facioscapulohumeral muscular dystrophy: epidemiological and molecular study in a north-east Italian population sample. Clin Genet. 2009;75:550–5.
Deenen JC, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJ, Bakker E, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056–9.
Padberg G. Facioscapulohumeral disease: University of Leiden; 1982. https://openaccess.leidenuniv.nl/handle/1887/25818.
Pastorello E, Cao M, Trevisan CP. Atypical onset in a series of 122 cases with facioscapulohumeral muscular dystrophy. Clin Neurol Neurosurg. 2012;114:230–4.
Nikolic A, Ricci G, Sera F, Bucci E, Govi M, Mele F, et al. Clinical expression of facioscapulohumeral muscular dystrophy in carriers of 1-3 D4Z4 reduced alleles: experience of the FSHD Italian National Registry. BMJ Open. 2016;6:e007798.
Tonini MM, Passos-Bueno MR, Cerqueira A, Matioli SR, Pavanello R, Zatz M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD). Neuromuscul Disord. 2004;14:33–8.
Ricci G, Scionti I, Sera F, Govi M, D’Amico R, Frambolli I, et al. Large scale genotype-phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain. 2013;136:3408–17.
Kilmer DD, Abresch RT, McCrory MA, Carter GT, Fowler Jr WM, Johnson ER, et al. Profiles of neuromuscular diseases. Facioscapulohumeral muscular dystrophy. Am J Phys Med Rehabil. 1995;74:S131–9.
Wohlgemuth M, van der Kooi EL, van Kesteren RG, van der Maarel SM, Padberg GW. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology. 2004;63:176–8.
Scully MA, Eichinger KJ, Donlin-Smith CM, Tawil R, Statland JM. Restrictive lung involvement in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2014;50:739–43.
de Greef JC, Lemmers RJLF, Camano P, Day JW, Sacconi S, Dunand M, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–54.
Lin F, Wang ZQ, Lin MT, Murong SX, Wang N. New insights into genotype-phenotype correlations in Chinese facioscapulohumeral muscular dystrophy: a retrospective analysis of 178 patients. Chin Med J (Engl). 2015;128:1707–13.
Statland JM, Tawil R. Risk of functional impairment in facioscapulohumeral muscular dystrophy. Muscle Nerve. 2014;49:520–7. This study is based on national FSHD registry data and provides important prospective, patient reported data over an average time span of 6 years.
Lunt PW, Jardine PE, Koch M, Maynard J, Osborn M, Williams M, et al. Phenotypic-genotypic correlation will assist genetic counseling in 4q35-facioscapulohumeral muscular dystrophy. Muscle Nerve. 1995;2:S103–S9.
Gaillard MC, Roche S, Dion C, Tasmadjian A, Bouget G, Salort-Campana E, et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology. 2014;83:733–42.
Larsen M, Rost S, El Hajj N, Ferbert A, Deschauer M, Walter MC, et al. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur J Hum Genet. 2015;23:808–16.
Calandra P, Cascino I, Lemmers RJ, Galluzzi G, Teveroni E, Monforte M, et al. Allele-specific DNA hypomethylation characterises FSHD1 and FSHD2. J Med Genet. 2016 [Epub ahead of print]
Padberg GW, van Engelen BG. Facioscapulohumeral muscular dystrophy. Curr Opin Neurol. 2009;22:539–42.
Lutz KL, Holte L, Kliethermes SA, Stephan C, Mathews KD. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology. 2013;81:1374–7.
Statland JM, Sacconi S, Farmakidis C, Donlin-Smith CM, Chung M, Tawil R. Coats syndrome in facioscapulohumeral dystrophy type 1: frequency and D4Z4 contraction size. Neurology. 2013;80:1247–50.
Statland JM, Donlin-Smith CM, Tapscott SJ, Lemmers RJ, van der Maarel SM, Tawil R. Milder phenotype in facioscapulohumeral dystrophy with 7-10 residual D4Z4 repeats. Neurology. 2015;85:2147–50.
Sacconi S, Lemmers RJ, Balog J, van der Vliet PJ, Lahaut P, van Nieuwenhuizen MP, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet. 2013;93:744–51. First description of an epigenetic modifier explaining disease variability in FSHD1. This opens up the possibility that mutations or variants in other genes important in chromatin regulation could explain clinical variability with kindreds with identical residual number of D4Z4 repeats.
Lemmers RJ, Goeman JJ, van der Vliet PJ, van Nieuwenhuizen MP, Balog J, Vos-Versteeg M, et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. 2015;24:659–69.
Bosnakovski D, Lamb S, Simsek T, Xu Z, Belayew A, Perlingeiro R, et al. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp Neurol. 2008;214:87–96.
Bosnakovski D, Xu Z, Gang EJ, Galindo CL, Liu M, Simsek T, et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. Embo J. 2008;27:2766–79.
Kowaljow V, Marcowycz A, Ansseau E, Conde CB, Sauvage S, Matteotti C, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord. 2007;17:611–23.
Snider L, Asawachaicharn A, Tyler AE, Geng LN, Petek LM, Maves L, et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet. 2009;18:2414–30.
Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell. 2012;22:38–51.
Yao Z, Snider L, Balog J, Lemmers RJ, Van Der Maarel SM, Tawil R, et al. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum Mol Genet. 2014;23:5342–52.
Pearson CM. Polymyositis. Annu Rev Med. 1966;17:63–82.
Dubowitz V, Brooke MH. Muscle biopsy; a modern approach. London: Saunders; 1973.
Bates D, Stevens JC, Hudgson P. “Polymyositis” with involvement of facial and distal musculature. One form of the fascioscapulohumeral syndrome? J Neurol Sci. 1973;19:105–8.
Munsat TL, Piper D, Cancilla P, Mednick J. Inflammatory myopathy with facioscapulohumeral distribution. Neurology. 1972;22:335–47.
Statland JM, Shah B, Henderson D, Van Der Maarel S, Tapscott SJ, Tawil R. Muscle pathology grade for facioscapulohumeral muscular dystrophy biopsies. Muscle Nerve. 2015;52:521–6.
DeVere R, Bradley WG. Polymyositis: its presentation, morbidity and mortality. Brain. 1975;98:637–66.
Brooke MH, Engel WK. The histologic diagnosis of neuromuscular diseases: a review of 79 biopsies. Arch Phys Med Rehabil. 1966;47:99–121.
Bethlem J. Myopathies. Amsterdam: North-Holland Publishing Co.; 1977.
Frisullo G, Frusciante R, Nociti V, Tasca G, Renna R, Iorio R, et al. CD8(+) T cells in facioscapulohumeral muscular dystrophy patients with inflammatory features at muscle MRI. J Clin Immunol. 2011;31:155–66.
Tasca G, Pescatori M, Monforte M, Mirabella M, Iannaccone E, Frusciante R, et al. Different molecular signatures in magnetic resonance imaging-staged facioscapulohumeral muscular dystrophy muscles. PLoS One. 2012;7:e38779.
Deidda G, Cacurri S, Piazzo N, Felicetti L. Direct detection of 4q35 rearrangements implicated in facioscapulohumeral muscular dystrophy (FSHD). J Med Genet. 1996;33:361–5.
Orrell RW, Tawil R, Forrester J, Kissel JT, Mendell JR, Figlewicz DA. Definitive molecular diagnosis of facioscapulohumeral dystrophy. Neurology. 1999;52:1822–6.
Jones TI, King OD, Himeda CL, Homma S, Chen JC, Beermann ML, et al. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin Epigenetics. 2015;7:37.
Attarian S, Salort-Campana E, Nguyen K, Behin A, Urtizberea JA. Recommendations for the management of facioscapulohumeral muscular dystrophy in 2011. Rev Neurol. 2012;168:910–8.
Tawil R, Kissel JT, Heatwole C, Pandya S, Gronseth G, Benatar M, et al. Evidence-based guideline summary: evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy. Neurology. 2015;85:357–64. These is the first attempt to develop evidence-based, standardized guidelines for the management of patients with FSHD.
Tawil R, McDermott MP, Pandya S, King W, Kissel J, Mendell JR, et al. A pilot trial of prednisone in facioscapulohumeral muscular dystrophy. FSH-DY Group. Neurology. 1997;48:46–9.
Munsat TL, Bradley WG. Serum creatine phosphokinase levels and prednisone treated muscle weakness. Neurology. 1977;27:96–7.
Elsheikh BH, Bollman E, Peruggia M, King W, Galloway G, Kissel JT. Pilot trial of diltiazem in facioscapulohumeral muscular dystrophy. Neurology. 2007;68:1428–9.
Kissel JT, McDermott MP, Mendell JR, King WM, Pandya S, Griggs RC, et al. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology. 2001;57:1434–40.
van der Kooi EL, Vogels OJ, van Asseldonk RJ, Lindeman E, Hendriks JC, Wohlgemuth M, et al. Strength training and albuterol in facioscapulohumeral muscular dystrophy. Neurology. 2004;63:702–8.
Passerieux E, Hayot M, Jaussent A, Carnac G, Gouzi F, Pillard F, et al. Effects of vitamin C, vitamin E, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: a double-blind randomized controlled clinical trial. Free Radic Biol Med. 2015;81:158–69.
Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–71.
Voet NB, van der Kooi EL, Riphagen II, Lindeman E, van Engelen BG, Geurts AC. Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst Rev. 2013;7:CD003907.
Andersen G, Prahm KP, Dahlqvist JR, Citirak G, Vissing J. Aerobic training and postexercise protein in facioscapulohumeral muscular dystrophy: RCT study. Neurology. 2015;85:396–403.
Abresch RT, Carter GT, Jensen MP, Kilmer DD. Assessment of pain and health-related quality of life in slowly progressive neuromuscular disease. Am J Hosp Palliat Care. 2002;19:39–48.
Jensen MP, Hoffman AJ, Stoelb BL, Abresch RT, Carter GT, McDonald CM. Chronic pain in persons with myotonic dystrophy and facioscapulohumeral dystrophy. Arch Phys Med Rehabil. 2008;89:320–8.
Lim JW, Snider L, Yao Z, Tawil R, Van Der Maarel SM, Rigo F, et al. DICER/AGO-dependent epigenetic silencing of D4Z4 repeats enhanced by exogenous siRNA suggests mechanisms and therapies for FSHD. Hum Mol Genet. 2015;24:4817–28.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Leo H. Wang declares no conflict of interest.
Rabi Tawil has received grants from the NIH and the FSH Society, consultancy fees from aTyr Pharma and Acceleron, and personal fees from Novartis.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Nerve and Muscle
Rights and permissions
About this article

Cite this article
Wang, L.H., Tawil, R. Facioscapulohumeral Dystrophy. Curr Neurol Neurosci Rep 16, 66 (2016). https://doi.org/10.1007/s11910-016-0667-0
Published:
DOI: https://doi.org/10.1007/s11910-016-0667-0