
Abstract
Although the vast majority of patients with status epilepticus (SE) respond fairly well to the first- or second-line anti-epileptics, a minority require anesthetic agents to put the seizures under control. An even smaller number of patients do not even respond to those and constitute the subgroup of super-refractory SE. Because of the small numbers, there are no definitive studies regarding its etiology, pathophysiology, and treatment, and those are still based on expert opinions. Encephalitides, either infectious, autoimmune, or paraneoplastic may be the main etiological factors. Induced pharmacological coma, immunosuppression, electrical brain stimulation, hypothermia, and ketamine are few of the newer but unproven therapeutic approaches that should be considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Status epilepticus (SE) was first described in the XXV–XXVI tablets of the Sakikku cuneiform written during the seventh or eighth century bc [1], but it was only until 1876 that SE was clinically defined by Bourneville [2]. Currently, SE is defined as one seizure lasting longer than 5 min or two or more seizures without returning to the neurological baseline in between. Refractory SE (RSE) is defined as ongoing seizures despite two appropriately selected and dosed anti-epileptic drugs (AEDs) including a benzodiazepine. More recently, a new term has gained popularity in the medical literature: super-refractory status epilepticus (SRSE). The term SRSE was first introduced during the London-Innsbruck Colloquium on SE held in Oxford in April 2011 and was defined as continuous or recurrent seizures lasting 24 h or more following initiation of anesthetic medications, including cases in which seizure control is attained after induction of anesthetic drugs but recurs on weaning the patient off the anesthetic agent [3].
SRSE has been typically, but not exclusively, described in two distinctive clinical situations: (1) in patients with severe acute brain injury and (2) in patients with no history of epilepsy who develop this condition with no overt cause. This latter situation has been considered by some to be a “syndrome” (entitled new-onset refractory status epilepticus (NORSE)) and was first described by Rathakrishnan and Wilder-Smith, Shorvon, and Khawaja et al. [4–6]. Other syndromes, such as febrile infection related epilepsy syndrome (FIRES) or devastating epileptic encephalopathy in school-aged children (DESC) [7] do not only introduce confusion about the entity and how it should be approached and treated but also challenge the reliability of available data about outcomes, risk factors, and the development of accurate prognostication tools.
Epidemiology of SRSE
The real incidence of SRSE is unknown. The description of several types of epileptic seizures has led to different classifications of SE. However, this can be simplified by referring to the electro-clinical features. Thus, SE could be classified by the presence and/or absence of motor convulsions into convulsive and non-convulsive status epilepticus (NCSE). These may be further subdivided into generalized and partial status. The accuracy of the diagnosis and clinical classification are important for the management and the occurrence of possible systemic complications. In the case of NCSE, there are two major groups. The first is the confused patient with behavioral changes and automatisms. The second type of NCSE patient encompasses those with brain injury that has led to decreased level of consciousness or coma. Convulsive SE can also transition to NCSE overtime.
It is estimated that SRSE is not uncommonly encountered in neuro-intensive care units, but its exact incidence, associated mortality, therapeutic strategy responses, and general outcome are not known. In one prospective study, 29/108 (22 %) of all the cases with status epilepticus admitted to the hospital failed to respond to first and second lines of therapy, and of these, 41 % (12 cases) required pharmacological coma induction. It is noteworthy that only 47 of the 108 patients had convulsive status epilepticus, and presumably, it is mainly in these in whom coma induction was required. It is also not clear how many of these coma-induced patients failed the emergence, meeting the definition of SRSE [8]. In a recent study conducted in a neuro-intensive care unit in a West China hospital from 2009 to 2012, a total of 98 patients were included. The percentage of NRSE, RSE, and SRSE were 67.3, 20.4, and 12.2 %, respectively [9]. Regarding SRSE, 67.7 % of the cases were in the setting of encephalitis and compared with a general SE mortality of 7 %, the mortality in the setting of SRSE reached 50 % [9]. In another retrospective study, 8-year data from 177 patients showed an incidence of SRSE at 16.9 %. Encephalitis was a most common etiology and constituted the most important factor to progress from non-refractory SE to SRSE [10].
Other retrospective studies have shown that 12–43 % of the cases with SE become refractory [11–14]. In a series of 35 patients, seven (20 %) recurred within 5 days of tapering; the anesthetic drug, and in all other studies, at least 50 % of those requiring anesthesia eventually became super refractory [11]. From these published findings, it can be estimated that approximately 10 to 15 % of all the cases of hospital-admitted SE will become super refractory at some point. However, the lack of prospective studies, the possible variation in type of treatment and time to initiation of therapy, the limited number of patients with this rare condition, and the possible selection bias of the retrospective studies, challenge our understanding of the incidence and epidemiology of SRSE.
Pathophysiology
One of the distinguishing characteristics of SE is the self-sustaining nature. Several animal models where seizures rapidly become self-sustaining despite the withdrawal of the epileptogenic stimulus have been developed and proved this hypothesis. Human data is limited. Some data can be extrapolated from De Lorenzo et al. study which reported that seizures lasting more than 30 min would rarely stop spontaneously compared with 47 % of those lasting between 10 and 29 min which would stop spontaneously without any intervention [15]. More recently, Jenssen et al. reported that no self-limited seizure lasted more than 11 min [16].
In the setting of SRSE, all the self-terminating mechanisms have failed and proved to be insufficient. In addition to the failure of mechanisms involved in seizure termination [17], there are several pathophysiologic processes ranging from changes in receptor configuration to genes expression that have been deemed responsible for perpetuation of SE. At cellular level, one of the most important findings has been the recognition of what has been called “receptor trafficking,” a concept first introduced by Arancibia and Kittler in 2009 [18]. Later, Smith and Kittler have described the highly dynamic state of receptor presence on the surface of axons and explained how receptors move onto (externalization), away from (internalization), and along the axonal membrane [19]. This “receptor trafficking” intensifies during SE, and the overall effect becomes a reduction in the number of functional γ-aminobutyric acid (GABA) receptors in the cells affected by the seizure discharge. As GABA is the principle inhibitory transmitter, this reduction in GABAergic activity may be an important reason for seizures to become persistent. Furthermore, the number of glutaminergic receptors at the cell surface increases and the reduction in the density of the GABA receptors is itself triggered by activation of the glutaminergic receptors. The reason for this shift at cellular level remains unknown. Although variability in genetic expression can explain it, the exact trigger is still a research subject. This loss of GABAergic receptor density is also the likely reason for the increasing ineffectiveness of GABAergic drugs (such as benzodiazepines or barbiturates) in controlling seizures as SE becomes more prolonged [20]. Moreover, it has been proposed that changes in the configuration of GABA-alpha receptor at the hippocampal level not only play a role in perpetuation of the epileptiform activity and subsequent development of SRSE, but it is also a reasonable explanation for the progressive loss of effectiveness of benzodiazepines in the treatment of SE.
Another contributing factor is the extracellular ionic environment, which can change in SE. For example, the normally inhibitory GABA(A)-mediated currents may become excitatory with changes in extracellular chloride concentrations [21].
This imbalance between inhibitory and excitatory circuitry is also important for long-term effects. The cerebral damage of SE includes neuronal cell necrosis, gliosis, and network reorganization. Excitotoxicity is the major player for cell death [22] and is being driven by a massive glutaminergic receptor overactivity. This would cause calcium influx into the cells leading to necrosis and apoptosis. The sequence of events leading to these outcomes can be initiated after few hours of continuous seizure activity. Thus, rapid instauration of therapy with anesthetics in the setting of SE is recommended because excitotoxicity could potentially be prevented by suppression of all electrographic activity and achieving EEG burst suppression [22]. Additional therapeutic strategies for SRSE based on the excitotoxicity hypothesis include, but are not limited to, hypothermia, barbiturates, steroids, and ketamine. The role of these interventions in the clinical outcomes is still a subject of debate and requires further research.
Mitochondrial failure has also been proposed as an alternative pathophysiologic mechanism for SRSE [23]. In 2002, Cock et al. postulated that mitochondrial insufficiency would lead to cell necrosis and apoptosis, leading to a maladaptive response and SE [24]. Inflammatory processes in the physiopathology of SRSE have also gained recognition [25]. In the study of 181 uncommon causes of SE identified from 588 articles, autoimmune disorders and inflammation were important etiologic factors for SE [25]. In this setting, the opening of the blood–brain barrier (BBB) is playing a major role in the perpetuation of seizures. The underlying mechanism is a maladaptive response of the astrocytes to the BBB damage, leading to activation of the innate immune system and disturbed homeostasis of the extracellular potassium and glutamate [26]. The role of inflammation in the etiology of SE could be also supported by the observed benefit of immunotherapy or immunomodulation in the treatment of status. More reports of RSE or SRSE have been recently published in the context of autoimmune or paraneoplastic encephalitis [27•, 28]. A vast and growing array of autoantibodies against intracellular and surface or synaptic neuronal targets leading to phenotypic variability in the spectrum of limbic encephalitis with or without refractory SE has been described in the context or not of malignancy and adds to the previous literature of Rassmussen’s encephalitis and Hashimoto’s encephalopathy [29]. The most common autoantibodies associated with seizures and SE include anti-Hu, anti-Ma2, anti-CV2/CRMP5, anti-Ri, ANNA3, anti-amphiphysin, anti-N-methyl-d-aspartate (NMDA) receptor, anti-LGI1 and CASPR2, anti-GABA-beta, anti-GluR3, and anti-mGluR5 [27•]. The diagnosis many times remains elusive, due to lack of knowledge, suspicion or plainly, lack of wide-spread availability of serologic testing or week-long delay for the results.
To these days, no genetic mechanism has been identified to explain the failure of seizure termination although it has been postulated that changes in gene expression occur and are in part responsible for the maintenance of the maladaptive response leading to SRES. It has been said that the changes in gene expression are the combined effect of repeated seizures, of seizure-induced neuronal death, and of the subsequent neuronal reorganization. The difficulty in consolidating the data pertaining to the role played by genes expression is partly explained by the inhibition of protein synthesis during SE as demonstrated by Wasterlain et al. [30].
Diagnosis
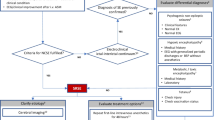
The diagnosis of convulsive SRSE is clinically obvious, but additional differential such as rigors due to sepsis, generalized dystonia, pseudostatus, tremors, and subcortical myoclonus must be taken into consideration. Non-convulsive SRSE may present with subtle clinical signs and be suspected in case of acute encephalopathy. For diagnosis, especially of the latter, and monitoring of the response to therapy, continuous electroencephalography with video capabilities is the best method. It is used to detect seizures and their localization, but this is not widely available in hospitals and ICUs. Serology for infectious, autoimmune, paraneoplastic agents, should be added to anti-epileptic drug levels (including free levels, when available), toxicology (including heavy metals), and genetic analysis (if positive family history). Lumbar puncture should be undertaken after neuroimaging (computed tomography and magnetic resonance imaging with and without contrast), excluding intracranial pathology and cerebrospinal fluid, which should be tested for infectious (viruses, parasites, fungi, and syphilis/borrelia) and paraneoplastic etiologies.
Treatment of SRSE
The development of well-established protocols for the treatment of SE and the rapid initiation of therapy are mandated in part by the deeper understanding of the pathophysiology of the condition and also by the existing SE-associated morbidity and mortality data. It has been estimated that mortality within 30 days after SE is about 7 to 39 % [11]. Morbidity, defined as severe focal neurological deficit, cognitive impairment, and development of epilepsy has been described in 3 to 13 % of cases [11]. As predicted, given the relatively recent introduction of SRSE in the medical literature [5], this information remains unknown for this condition and data can only be extrapolated from previous experiences gained in the treatment of SE and from the limited information directly applicable to SRES patients obtained from small case series and/or case reports. In general, the treatment goals for SE include primarily control of seizures and therefore avoidance of excitotoxicity, their recurrence, and systemic complications. In SRES, where the outcome may be even worse than that in SE, the treatment goals in general are the same, except that avoidance of excitotoxicity is no longer applicable, as in this setting, excitotoxicity would have been already ongoing. The possible implications of the theoretically ongoing excitotoxicity in clinical outcomes and effectiveness of the selected therapy remain to be seen.
SRSE is treated in intensive care units. It is unknown if admission to a neuro-intensive care unit entails better outcomes than admission to general ICUs [31]. The therapeutic approach generally includes the use of assisted ventilation and full cardiovascular monitoring. Table 1 presents a suggested path for treating RSE and SRSE, with a transition between the two (stages 1 to 2). It is generally accepted that general anesthesia is required and constitutes the central pillar of treatment. However, questions about the choice of anesthetic agent, duration of therapy, combination of anti-epileptic drugs (AEDs), and effect of other treatment modalities to the cessation of seizures and the clinical outcomes still remain.
AEDs are traditionally given concomitantly with anesthetics for the treatment of SRSE. The idea is to prevent seizures after anesthetics have been discontinued which usually occurs 24 to 48 h after instauration of therapy. However, the precise role and effectiveness of different AEDs in the setting of SRSE is unknown [5, 8]. There are no randomized trials comparing the different AEDs for SRSE, which sharply contrasts the number of studies describing their effectiveness at the onset of seizures or SE.
Barbiturate anesthesia using thiopental or pentobarbital has been widely accepted. Barbiturates act by enhancing the action of GABA-alpha receptor. Additionally, it has been postulated that by lowering core temperature, these agents may exert a neuroprotective role that may be beneficial in SRSE. The main disadvantages of this pharmacological family is their rapid redistribution leading to accumulation and prolonged half-life that can reach hours or days and thus a long sedative effect in a patient with a potentially already compromised neuro-exam, which needs, nonetheless, to be assessed frequently. Other important side effects are the cardiorespiratory depression and hypotension, the respiratory depression and need for full ventilator support, and the ileus mandating parenteral nutrition [32, 33•]. The depth and duration of the EEG suppression that must be achieved by barbiturates is unknown. Some experts recommend, instead of burst-suppression pattern, complete suppression or “flat record” because of better seizure control and fewer relapses [34]. The same group showed that patients with more prolonged barbiturate treatment (>96 h) and those receiving phenobarbital at the time of pentobarbital taper were less likely to relapse [35]. In a recent reviews, it was found that barbiturates control refractory and super-refractory SE in 64 % of the patients and were ineffective and unable to control seizures in only 5 % [8, 36•]. However, the outcome in these cases may still be poor. At 1 year post-discharge, 74 % are dead or in a state of unresponsive wakefulness, 16 % severely disabled, and only 10 % have no or minimal disability [37].
Midazolam is another anesthetic agent widely used for the treatment of RSE and SRSE. It rapidly enters the brain tissue and exerts a powerful short-duration action without the risk of accumulation. Its mechanism of action is biding and enhancing the action of GABA-alpha receptor. Its main advantage is its potent anti-epileptic effect. The main disadvantage is its tendency for developing tolerance and leading to seizure recurrence. Singhi et al. [38] and Morrison et al. [39] reported the occurrence of breakthrough seizures in 47 to 57 %. More recently, Ferlisi et al. reported only a 3 % incidence of breakthrough seizures, with less than 1 % withdrawal seizures and therapy failure due to side effects. In the same retrospective analysis, midazolam was able to control seizures in 78 %, but no control was achieved in 16 % of patients. Mortality using this therapy was reported at 2 % [8, 36•].
Propofol is another anesthetic agent routinely used for the treatment of RSE and SRSE. It is believed that its main action is achieved through modulation of GABA-alpha receptor as with the previously mentioned agents. Its main advantage is its rapid onset and recovery after infusion. The main disadvantage is the risk for developing propofol infusion syndrome (PRIS) [40], which is more prevalent in the pediatric population and in patients concomitantly treated with steroids or cathecholamines. Another side effect is drug-induced involuntary movements that can resemble seizures. It has been suggested that the occurrence of these movements is due to the lack of cortical inhibition or may be peripheral in nature. In a retrospective analysis, propofol led to 68 % seizure control in the setting of RSE and SRSE, with a failure of therapy of 11 % and occurrence of breakthrough seizures and withdrawal seizures of 1 and 6 %, respectively [36•].
A unique feature in the progression toward SE and SRSE is the time-dependent development of pharmacoresistance. This has been described with benzodiazepines, which act on GABA-alpha receptors and may decrease their potency by 20-fold in 30 min [41]. It is important to highlight that GABA-alpha receptor is the target shared by the previously mentioned anesthetics. By contrast, NMDA receptor blockers continue to be effective in stopping seizures at least in animal models [42]. This observation has led to the use of ketamine for RSE and SRSE. Ketamine is an anesthetic agent that possesses a dual effect by biding on the GABA-alpha receptor and antagonizing the N-methyl-d-aspartate receptor. Its main advantage is the lack of cardiac depressant properties. In a retrospective study of 58 patients, ketamine was likely responsible for seizure control in 12 % and possibly responsible in an additional 20 %. No responses were observed when infusion rate was lower than 0.9 mg kg−1 h−1 or when ketamine was introduced at least 8 days after onset of SE or after failure of seven or more drugs [43]. Late development of brain atrophy possibly due to excitotoxicity caused by the drug mandates caution [44].
The use of inhalational halogenated anesthetics has been reported in 11 cases [45]. However, difficulties providing treatment in the ICU setting and their associated systemic complications [46] make these a less desirable therapeutic option. Isoflurane, however, can be considered in cases of acute intermittent porphyria causing SRSE [47].
Intravenous magnesium is the drug of choice for the treatment of seizures in eclampsia [48]. It is believed that its anti-epileptic effect is achieved by blocking NMDA receptors. Magnesium’s lifesaving effect in acquired hypomagnesemia and eclampsia makes it an attractive option in the treatment of SRSE, but data to support its use are scarce. Its effectiveness has been reported in three cases, two of them with mitochondrial disease. Infusion rates ranges from 2 to 6 g/h, but levels higher than 8 mEq/L may risk cardiovascular collapse and should be avoided [49].
Pyridoxine (B6) use has been suggested for the treatment of SRSE [32]. Pyridoxal phosphate, its activated form, is a coenzyme in the conversion of glutamic acid to GABA by the enzyme glutamic acid decarboxylase. Its indication and effectiveness in SRSE remain unknown. However, its use may be supported by reported cases of acquired pyridoxine deficiency, one during pregnancy and the other secondary to malnourishment, as well after isoniazid intoxication (this drug inhibits the enzyme pyridoxine phosphokinase, which transforms pyridoxine to pyridoxal phosphate). The infusion of pyridoxine (100 mg/day or up to 1 g IV pyridoxine for each gram isoniazid injested [50]) carries no major risk if administered for few days and therefore its use during SRSE may be justified.
Hypothermia has been reported in four adult patients with SRSE [51]. Temperatures of 30–35 °C were achieved for 20–61 h using endovascular cooling catheters. It is unclear if hypothermia had an effect since these patients were also receiving barbiturates and benzodiazepines, but after rewarming, all had reduction of seizure frequency and two became seizure free. This is not a benign treatment and should be used in ICUs with experience in its use for other conditions (usually post cardiac arrest). Acid–base disturbances, arrhythmias, coagulopathy, and bowel ischemia can occur [52].
Various forms of electric stimulation of the seizing brain have been reported in case reports or small case series of SRSE with varying degrees of success. What remains unclear is which brain areas should be stimulated and what parameters should be used [53]. A 26-year-old woman was treated with low-frequency cortical stimulation via subdural electrodes for seven consecutive days. Previously, she was on two anesthetics and high doses of two to four enteral AEDs. She responded after 1 day of stimulation, and one anesthetic agent was successfully discontinued. Seizures only returned by the 4th day when the second anesthetic had been reduced by 60 % [54]. In another case, a 30-year-old man with SRSE had left vagal nerve stimulator placement after not responding to pentobarbital coma for 9 days. On the following day, EEG revealed resolution of previously observed periodic lateral epileptiform discharges and he became seizure free [55]. Electroconvulsive therapy (3 sessions/week, 6 total) was administered in a patient who was in SRSE and not responding to pentobarbital coma for 40 days. After the 2nd session, the barbiturate was removed and eventually the patient recovered within 1 month [56]. Low-frequency repetitive transcranial magnetic stimulation has been used in a patient with focal SRSE. Stimulation was delivered over the epileptogenic focus in 1-h sessions daily for 8 days with electroencephalographic and clinical improvement [57].
The decision to start blindly immunotherapy for suspected autoimmune SRSE is difficult. One should remember that these cases are treatment resistant and no randomized trials have been published. When no other etiology has been found, such an approach should be considered after a paraneoplastic antibody panel has been collected (serum or CSF), even in the waiting period before the results are back. Higher clinical suspicion for autoimmune SE should be present when no longstanding history of epilepsy is reported, when prominent memory loss and psychiatric symptomatology is quickly evolving, and when a known malignancy is present and other neurological signs, such as ataxia or autonomic dysfunction, coexist [27•]. If on CT or PET of the entire body a tumor is discovered, then resection of the mass may improve seizure control. The AEDs are the same as those used against SRSE. Many patients in SE already host infections, and immunosuppressive treatments should be initiated only after the infection is under control. In parallel with the AEDs, high-dose corticosteroids (1 g methylprednisolone IV for 5 days, followed by 1 mg kg−1 prednisone day−1) with or without intravenous immunoglobulin (2 g/kg over 5 days) or plasmapheresis (five sessions) are treatments that can be used based on expert opinions and anecdotal experience. Second-line treatments include cyclophosphamide or rituximab and, for maintenance or recurrences, mycophenolate mofetil or azathioprine. Surgical resection of an epileptogenic focus or for Rassmussen’s encephalitis should also be considered [27•].
Conclusion
SRSE is likely due to rare conditions, usually encephalitides, autommune, or paraneoplastic syndromes, being itself rare. If general anesthetics fail to control the seizures or their recurrence after tapering the dose, various other therapeutic options should be tried, based on scanty data and unclear effect on outcomes.
References
Papers of particular interest published recently have been highlighted as: • Of importance
Chen JW, Naylor DE, Wasterlain CG. Advances in the pathophysiology of status epilepticus. Acta Neurol Scand Suppl. 2007;186:7–15.
Bourneville DM (1876) L’etat de mal epileptique. In: Recherches cliniques et therapeutiques sur l’epilepsie et l’hysterie. Compte-rendu des observations recueillies a la Salpetriere. Delahaye, Paris
Hocker S, Tatum WO, LaRoche S, Freeman WD. Refractory and super-refractory status epilepticus—an update. Curr Neurol Neurosci Rep. 2014;14:452.
Rathakrishnan R, Wilder-Smith EP. New onset refractory status epilepticus (NORSE). J Neurol Sci. 2009;284:220. author reply 20–1.
Shorvon S. Super-refractory status epilepticus: an approach to therapy in this difficult clinical situation. Epilepsia. 2011;52 Suppl 8:53–6.
Khawaja AM, DeWolfe JL, Miller DW, Szaflarski JP. New-onset refractory status epilepticus (NORSE)—the potential role for immunotherapy. Epilepsy Behav. 2015;47:17–23.
Ismail FY, Kossoff EH. AERRPS, DESC, NORSE, FIRES: multi-labeling or distinct epileptic entities? Epilepsia. 2011;52:e185–9.
Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain. 2011;134:2802–18.
Tian L, Li Y, Xue X, et al. Super-refractory status epilepticus in West China. Acta Neurol Scand. 2015;132:1–6.
Jayalakshmi S, Ruikar D, Vooturi S, et al. Determinants and predictors of outcome in super refractory status epilepticus—a developing country perspective. Epilepsy Res. 2014;108:1609–17.
Holtkamp M, Othman J, Buchheim K, Masuhr F, Schielke E, Meierkord H. A “malignant” variant of status epilepticus. Arch Neurol. 2005;62:1428–31.
Lowenstein DH, Alldredge BK. Status epilepticus at an urban public hospital in the 1980s. Neurology. 1993;43:483–8.
Mayer SA, Claassen J, Lokin J, Mendelsohn F, Dennis LJ, Fitzsimmons BF. Refractory status epilepticus: frequency, risk factors, and impact on outcome. Arch Neurol. 2002;59:205–10.
Rossetti AO, Logroscino G, Bromfield EB. Refractory status epilepticus: effect of treatment aggressiveness on prognosis. Arch Neurol. 2005;62:1698–702.
DeLorenzo RJ, Garnett LK, Towne AR, et al. Comparison of status epilepticus with prolonged seizure episodes lasting from 10 to 29 minutes. Epilepsia. 1999;40:164–9.
Jenssen S, Gracely EJ, Sperling MR. How long do most seizures last? A systematic comparison of seizures recorded in the epilepsy monitoring unit. Epilepsia. 2006;47:1499–503.
Lado FA, Moshe SL. How do seizures stop? Epilepsia. 2008;49:1651–64.
Arancibia-Carcamo IL, Kittler JT. Regulation of GABA(A) receptor membrane trafficking and synaptic localization. Pharmacol Ther. 2009;123:17–31.
Smith KR, Kittler JT. The cell biology of synaptic inhibition in health and disease. Curr Opin Neurobiol. 2010;20:550–6.
Macdonald RL, Kapur J. Acute cellular alterations in the hippocampus after status epilepticus. Epilepsia. 1999;40 Suppl 1:S9–20. discussion S21-2.
Lamsa K, Taira T. Use-dependent shift from inhibitory to excitatory GABAA receptor action in SP-O interneurons in the rat hippocampal CA3 area. J Neurophysiol. 2003;90:1983–95.
Meldrum B. Excitotoxicity and epileptic brain damage. Epilepsy Res. 1991;10:55–61.
Cock H. The role of mitochondria in status epilepticus. Epilepsia. 2007;48 Suppl 8:24–7.
Cock HR, Tong X, Hargreaves IP, et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res. 2002;48:157–68.
Tan RY, Neligan A, Shorvon SD. The uncommon causes of status epilepticus: a systematic review. Epilepsy Res. 2010;91:111–22.
Friedman A, Dingledine R. Molecular cascades that mediate the influence of inflammation on epilepsy. Epilepsia. 2011;52 Suppl 3:33–9.
Lopinto-Khoury C, Sperling MR. Autoimmune status epilepticus. Curr Treat Options Neurol. 2013;15:545–56. A well written synopsis of autoimmune SE with a Diagram incorporating diagnosis and treatment.
Ramanathan S, Wong CH, Rahman Z, Dale RC, Fulcher D, Bleasel AF. Myoclonic status epilepticus as a presentation of caspr2 antibody-associated autoimmune encephalitis. Epileptic Disord. 2014;16:477–81.
Davis R, Dalmau J. Autoimmunity, seizures, and status epilepticus. Epilepsia. 2013;54 Suppl 6:46–9.
Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993;34 Suppl 1:S37–53.
Varelas PN, Corry J, Rehman M, et al. Management of status epilepticus in neurological versus medical intensive care unit: does it matter? Neurocrit Care. 2013;19:4–9.
Varelas PN. How I, treat status epilepticus in the Neuro-ICU. Neurocrit Care. 2008;9:153–7.
Varelas PN, Spanaki MV, Mirski MA. Status epilepticus: an update. Curr Neurol Neurosci Rep. 2013;13:357. A broader recent review of SE, encorporating responsive and refractory status.
Krishnamurthy KB, Drislane FW. Depth of EEG suppression and outcome in barbiturate anesthetic treatment for refractory status epilepticus. Epilepsia. 1999;40:759–62.
Krishnamurthy KB, Drislane FW. Relapse and survival after barbiturate anesthetic treatment of refractory status epilepticus. Epilepsia. 1996;37:863–7.
Ferlisi M, Shorvon S. The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapy. Brain. 2012;135:2314–28. The most comprehensive review in the topic of SRSE.
Pugin D, Foreman B, De Marchis GM, et al. Is pentobarbital safe and efficacious in the treatment of super-refractory status epilepticus: a cohort study. Crit Care. 2014;18:R103.
Singhi S, Murthy A, Singhi P, Jayashree M. Continuous midazolam versus diazepam infusion for refractory convulsive status epilepticus. J Child Neurol. 2002;17:106–10.
Morrison G, Gibbons E, Whitehouse WP. High-dose midazolam therapy for refractory status epilepticus in children. Intensive Care Med. 2006;32:2070–6.
Smith H, Sinson G, Varelas P. Vasopressors and propofol infusion syndrome in severe head trauma. Neurocrit Care. 2009;10:166–72.
Mazarati AM, Baldwin RA, Sankar R, Wasterlain CG. Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Res. 1998;814:179–85.
Mazarati AM, Wasterlain CG. N-methyl-d-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neurosci Lett. 1999;265:187–90.
Gaspard N, Foreman B, Judd LM, et al. Intravenous ketamine for the treatment of refractory status epilepticus: a retrospective multicenter study. Epilepsia. 2013;54:1498–503.
Ubogu EE, Sagar SM, Lerner AJ, Maddux BN, Suarez JI, Werz MA. Ketamine for refractory status epilepticus: a case of possible ketamine-induced neurotoxicity. Epilepsy Behav. 2003;4:70–5.
Kofke WA, Young RS, Davis P, et al. Isoflurane for refractory status epilepticus: a clinical series. Anesthesiology. 1989;71:653–9.
Mirsattari SM, Sharpe MD, Young GB. Treatment of refractory status epilepticus with inhalational anesthetic agents isoflurane and desflurane. Arch Neurol. 2004;61:1254–9.
Payne TA, Bleck TP. Status epilepticus. Crit Care Clin. 1997;13:17–38.
Gillon TE, Pels A, von Dadelszen P, MacDonell K, Magee LA. Hypertensive disorders of pregnancy: a systematic review of international clinical practice guidelines. PLoS One. 2014;9:e113715.
Smith JM, Lowe RF, Fullerton J, Currie SM, Harris L, Felker-Kantor E. An integrative review of the side effects related to the use of magnesium sulfate for pre-eclampsia and eclampsia management. BMC Pregnancy Childbirth. 2013;13:34.
Alvarez FG, Guntupalli KK. Isoniazid overdose: four case reports and review of the literature. Intensive Care Med. 1995;21:641–4.
Corry JJ, Dhar R, Murphy T, Diringer MN. Hypothermia for refractory status epilepticus. Neurocrit Care. 2008;9:189–97.
Motamedi GK, Lesser RP, Vicini S. Therapeutic brain hypothermia, its mechanisms of action, and its prospects as a treatment for epilepsy. Epilepsia. 2013;54:959–70.
Walker MC. The potential of brain stimulation in status epilepticus. Epilepsia. 2011;52 Suppl 8:61–3.
Schrader LM, Stern JM, Wilson CL, et al. Low frequency electrical stimulation through subdural electrodes in a case of refractory status epilepticus. Clinical Neurophysiol. 2006;117:781–8.
Patwardhan RV, Dellabadia Jr J, Rashidi M, Grier L, Nanda A. Control of refractory status epilepticus precipitated by anticonvulsant withdrawal using left vagal nerve stimulation: a case report. Surg Neurol. 2005;64:170–3.
Carrasco Gonzalez MD, Palomar M, Rovira R. Electroconvulsive therapy for status epilepticus. Ann Intern Med. 1997;127:247–8.
Thordstein M, Constantinescu R. Possibly lifesaving, noninvasive, EEG-guided neuromodulation in anesthesia-refractory partial status epilepticus. Epilepsy Behav. 2012;25:468–72.
Compliance with Ethics Guidelines
Conflict of Interest
Mauricio Ruiz Cuero declares no conflict of interest. Panayiotis N. Varelas reports personal fees (Advisory Board) from UCB and grants from SAGE Therapeutics.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Critical Care
Rights and permissions
About this article

Cite this article
Cuero, M.R., Varelas, P.N. Super-Refractory Status Epilepticus. Curr Neurol Neurosci Rep 15, 74 (2015). https://doi.org/10.1007/s11910-015-0594-5
Published:
DOI: https://doi.org/10.1007/s11910-015-0594-5