
Abstract
Mutations in LRRK2 are associated with inherited Parkinson’s disease (PD) in a large number of families, and the genetic locus containing the LRRK2 gene contains a risk factor for sporadic PD. The LRRK2 protein contains several domains that suggest a role in cellular signaling, including a kinase domain. It is also clear that LRRK2 interacts, either physically or genetically, with several other important proteins implicated in PD, suggesting that LRRK2 may be a central player in the pathways that underlie parkinsonism. As such, LRRK2 has been proposed to be a plausible target for therapeutic intervention, with kinase inhibition being pursued most actively. However, there are still several fundamental aspects of LRRK2 biology and function that remain unresolved at this time. This review will focus on the key questions of normal function of LRRK2 and how this might be related to the pathophysiology of PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Many discussions about the etiology of Parkinson’s disease (PD) start from the premise that the set of causes of this condition include genetic and environmental factors, but I am not convinced that this statement is correct. The replicated and known causal factors for PD are the following: genes, in the form of segregating variants in a modest but appreciable number of families and associated loci in sporadic disease, and aging, which is a required factor as no one is, to my knowledge, born with PD. Between age and genetics, one can explain the majority of risk in familial PD and about one third of lifetime risk in sporadic cases.
Why make this point? Because the diseases most likely to be solved are those where the etiology is best understood—vaccination against infectious disease being perhaps the most striking example—and so it is important to have clarity around causation. However, some humility is helpful, as understanding the genetic basis of disease does not immediately lead to cures. In some cases, there are spectacular successes, such as the use of high-dose riboflavin treatment in Brown-Vialetto-Van Laere syndrome caused by mutations in a riboflavin transporter [1]. However, in other cases, there is a distinct lag between finding a mutation and developing rational therapies, such as with cystic fibrosis [2]. In the case of PD, the first clearly defined causal gene was identified in 1997 [3] but no drug that affects the underlying disease progression is yet available.
In this context, the case of LRRK2 merits particular discussion. As will be discussed below, LRRK2 has attracted a great deal of interest as a potential drug target for PD, and yet, how it causes disease is poorly understood. My aim with this review is largely to discuss what is known and what is not known about LRRK2 with the underlying assumption that better understanding of how mutations affect the biology of the protein will identify novel targets for therapies.
LRRK2 as a Causative Gene and as a Risk Modifier
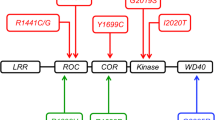
Mutations in LRRK2 were identified in a series of families from many different parts of the world [4–6]. The domain organization of LRRK2 and where the major mutations are located within that structure are shown in Fig. 1. All families show autosomal dominant inheritance of parkinsonism with an age-dependent but incomplete penetrance—there are individuals who live until old age without any clinical signs of PD [7]. In contrast to many other forms of parkinsonism that are generally rare, some LRRK2 mutations are relatively common. For example, the G2019S variant is present worldwide [8] but has a higher prevalence in people in North Africa where it may account for 30–40 % of all PD cases [9]. It is worth noting that many of these cases are apparently sporadic, probably due to low penetrance [10]. Because LRRK2 mediated disease requires aging, and even in older age not all carriers of LRRK2 express the disease phenotype, it is possible that relatives of a given index case may not have had PD within their lifetime.

LRRK2, mutations, regulation, and interactions. The domain structure of LRRK2 is shown in the center of the figure (blue) and contains (from N- to C-termini) leucine-rich repeats (LRR), a Ras of complex protein (ROC) and C-terminal of ROC (COR) bidomain, a kinase domain, and a WD40 domain. Above the diagram are indications of the biochemical functions of each region and, above those, are some known regulators of phosphorylation status. Below the outline are the major Mendelian mutations (red) and one of the more common risk factors (orange) and an indication of their effects on the measurable biochemical activities of LRRK2. Finally, in the lower portion of the figure, some of the more reliable protein interactions are shown; all of these have been reported in more than one paper and with more than one technique used apart from GAK (cyclin G-associated kinase) which has to date only been reported once. This is included because it is one of two candidate risk factor genes identified by GWAS. Two additional genes that are important for sporadic PD risk are a-synuclein and tau, which have not yet been shown to bind directly to LRRK2 but do appear to be functionally related to some of the pathways in which LRRK2 is involved
All of the above variants that cause disease in families are non-synonymous point mutations, but there is an additional level of variation that was identified by genome-wide association studies (GWAS). The principle of GWAS is that a large collection of people without or with disease, in this instance sporadic PD, is genotyped at markers spread over all chromosomes. These markers are picked because they have more than one variant that is relatively common (usually more than 5 % of a population), but not because they are targeted to a gene that we suspect might be involved in a given disease. It was therefore hugely surprising that in the list of risk factors for sporadic PD nominated by GWAS, the region on chromosome 12 that harbors LRRK2 was identified and subsequently replicated across several studies [11–14].
LRRK2 is therefore a candidate for a pleomorphic risk locus [15], i.e., a genetic region that contains both Mendelian variants and sporadic risk factors. Formally, it is not certain that LRRK2 itself is the gene that causes risk, as GWAS nominate large regions not specific variants in single genes. However, given the known pathogenic role of LRRK2 in familial PD, parsimony suggests that LRRK2 is the most likely candidate at this locus. It is not known how variants around LRRK2 influence the risk of disease. Amino acid changes are unlikely to be important as the gene has been sequenced extensively and most variants are too rare to explain the signal common enough to be picked up in GWAS. More likely, changes in expression levels or splicing of LRRK2 contribute to disease risk. An important question is whether higher or lower levels of LRRK2 activity are associated with disease pathogenesis. I will next discuss what the balance of evidence is for loss and gain of function of LRRK2 in relation to disease.
Mutant LRRK2 Alleles Produce Biochemically Active Proteins
Early experiments performed after the initial cloning of LRRK2 were aimed at a fundamental characterization of the active enzyme. LRRK2 has a clearly defined kinase sequence and a Ras of complex protein (ROC) domain, predicted on the basis of homology to bind and hydrolyze GTP (Fig. 1). Intervening the ROC and kinase domains is a C-terminal of ROC (COR) domain. Outside of the catalytic core are a series of repeats including the leucine-rich repeats (LRR) that give LRRK proteins their name and a C-terminal WD40 domain. Thus, it was initially predicted that LRRK2 would have kinase and GTPase activities in common with other members of the ROCO family of proteins [16].
Early results confirmed LRRK2 as an active kinase and reported an ∼twofold increase with the G2019S mutation [17, 18]. Other mutations either had only a modest increase in activity or were similar to wild-type protein. This result has been confirmed across many independent studies using different ways to measure kinase activity [19]. That the increase in activity with G2019S is seen no matter how LRRK2 is assayed suggests that the enhanced function is an inherent property of the mutant protein and that disease is not caused by a loss of kinase activity. That disease is associated with an enhanced function would then mean that inhibition of kinase activity should block toxicity, leading to a new therapeutic approach for PD.
Results comparing kinase-active and kinase-dead constructs in cell [18, 20] and mouse models [21] suggest that kinase activity is indeed required for detrimental effects of mutant LRRK2. Because of these results, several groups have developed small molecules that will compete for ATP in the kinase domain of LRRK2 and inhibit activity [22–25] and some LRRK2 inhibitors block neurodegeneration in vivo [26].
Despite these positive results, it is unclear if LRRK2 kinase inhibitors could be therapeutically useful. Dosing primates with inhibitors can cause lung pathology similar to that seen in knockout mice [27]. The similarity between phenotypes suggests that the toxic effects might be mediated through LRRK2 itself, raising a potential safety concern for LRRK2 kinase inhibitors in general. Additionally, not all mutations in LRRK2 increase kinase activity and at least one risk factor variant, G2385R, decreases kinase activity even if placed in a construct also including the hyperactive G2019S mutation [28]. There are several ways to interpret this data, but two obvious possibilities are that either (a) both increased and decreased activities are associated with disease risk or (b) disease mechanisms do not necessarily involve the kinase activity of LRRK2 [29]. Also, in some assays, LRRK2 inhibition can make the wild-type protein behave like mutant forms. LRRK2 is phosphorylated at a series of residues around the LRR region including S910, S935, S955, and S973 that are important for binding 14-3-3 proteins [30, 31]. Phosphorylation at these residues is controlled by combined action of kinases, which might include casein-kinase 1 [32] and IKK family members [33], and phosphatases including PP1a [34]. Mutant forms of LRRK2 tend to be dephosphorylated [30, 31], and inhibitors either of upstream kinases [32] or LRRK2 itself [35] mimic this effect. The mechanistic details of why LRRK2 inhibitors cause this effect are not resolved, but the data show that inhibited LRRK2 might behave like the pathogenic form, at least in these biochemical assays, leaving the possibility that such compounds might have detrimental effects open. Finally, it was not widely noted in earlier papers but several kinase-inactive versions of LRRK2 are less stable than kinase-active versions especially in neurons. Thus, some of the “protective” effects of kinase inactivation might be explained by decreased protein levels, even if the initial results were correct [36]. At this time, there are therefore several reasons to think that kinase inhibition of LRRK2 might be therapeutically useful for PD but enough uncertainty to consider whether other portions of the molecule might also be targeted [29].
In turn, this leads to the question of what the other regions of LRRK2 do. As mentioned above, the ROC-COR domain of LRRK2 was predicted to bind and hydrolyze GTP as a member of the ROCO protein family [37] and possibly part of the mechanistic group of G proteins that are regulated by dimerization [38]. Several groups were able to show that LRRK2 can bind GTP and that mutations do not seem to affect binding affinity [39–41]. Although GTPase activity of LRRK2 is low compared to small GTPases like Rab or Ras, it has been measured and a consistent result is that mutations in the ROC or COR domains are associated with lower GTPase activity, i.e., slower rates of turnover from GTP to GDP [39, 40, 42–44].
To my mind, the interpretation of these results depends on whether the GTP- or GDP-bound form of LRRK2 is the “active” cellular complex. If, like many of the small GTPases, GTP-LRRK2 were active, then, lower GTPase activity would be associated with longer time in the activated state. Thus, a diminished biochemical activity might lead to a higher cellular activity by virtue of having a persistent function. This view, which is admittedly speculative at this time, would then support the concept that ROC/COR LRRK2 mutations, like kinase mutations, are biochemically active.
Although the data therefore now supports the original proposal that LRRK2 has both kinase and GTPase activity, there are a few areas that remain unclear. First, while LRRK2 can be assayed as a kinase, the true substrate for LRRK2 remains uncertain with many proposed but few being proven unambiguously at physiological levels [45]. A convenient way to assay LRRK2 is to monitor autophosphorylation, and there is one site at S1292 that can be measured in vivo [25], so it remains possible if unlikely that there are no actual external substrates. Second, because of the difficulties in measuring GTPase activity in vitro, it is uncertain whether LRRK2 can promote the turnover of GTP to GDP in the cell without additional regulators. Some models have proposed that their GTPase accessory proteins (GAPS) and GTP exchange factors (GEFs) can bind LRRK2, specifically ArfGap1 [42, 46] and ARHGEF7 [32, 47]. However, in other models, LRRK2 dimerization, which has been shown to occur in the full-length protein by several groups [48–51], has been proposed to control GTPase activity without the requirement for accessory proteins [38]. The dimerization motif of some LRRK2 homologues is the COR domain [52], potentially explaining how COR mutations diminish GTPase activity of LRRK2. If correct, then, this model might explain why activity is hard to measure—protein concentration of LRRK2 in the assay would have to be high enough to support dimer formation in the test tube, which is difficult to obtain for such a large protein.
Thirdly, how GTPase and kinase activities relate to each other is not resolved. Intuitively, the presence of two distinct activities in the same molecule has been selected evolutionarily because they require tight spatial or temporal coordination. The initial model was that, in the way that small GTPases like Ras regulate kinases like Raf, GTP binding to the ROC/COR region of LRRK1 or LRRK2 stimulates kinase activity [41, 53]. However, subsequent studies found that while mutations that diminish GTP/GDP binding capacity of LRRK2 do indeed have lower kinase activity, adding excess non-hydrolyzable GTP homologues does not increase kinase activity as predicted [54, 55]. Therefore, the available data does not support the simple model that GTP-bound LRRK2 has more kinase activity.
Although this might seem a rather small point, it has an important implication for understanding mutations in LRRK2, which can be illustrated by the following logic. If mutations either increase kinase activity (G2019S) or diminish GTPase activity (R1441C, Y1699C) and if the GTP-bound form of LRRK2 has higher kinase activity than GDP-LRRK2, then, all mutations have the same biochemical output of more kinase activity. Importantly, for the discussion here, this model has all mutant alleles producing biochemically active protein. However, some mutations do not fit in this simple rubric—G2385R as mentioned above has lower kinase activity, and I2020T has variable effects on kinase activity depending on the assay used [56]. Hence, we cannot at this time show that all mutations affect the same biochemical outputs of the protein.
I think that this is a question that is in danger of being left to one side with the mistaken impression that the answer is either known or uninteresting. I do think it is likely that all mutations lead to the same output at a cellular level that, from there, impact the same pathogenic process and some candidate examples will be given below. However, there are several ways to resolve the available biochemical data with the concept of a unified pathogenic mechanism. Perhaps we are thinking about “activity” in a rather simple steady state manner when in fact considering the dynamics and sequence of events might be more helpful. For example, LRRK2 is a dimer [50] that is proposed to be more kinase active than monomer when associated with cellular membranes [48, 57]. It is therefore possible that the “activity” of LRRK2 is regulated by a combination of monomer to dimer transitions, in turn dependent on concentration, and/or localization within the cell. If such a scenario were correct, then, we would simply revise the original models to include a temporal and spatial component.
An alternative view is that the kinase and GTPase activities are not relevant because LRRK2 mutations are pathogenic by virtue of a loss of normal function. This would be consistent with LRRK2 being both a risk factor and a Mendelian gene, so long as the risk factor variants are associated with low expression. However, I think this is an unlikely explanation. While there are phenotypes associated with both loss of function and PD-mutant alleles in mice, they tend to be different quantitatively or qualitatively. For example, there are strong kidney phenotypes in LRRK2 knockout mice that are not shared when G2019S is knocked in [58]. Overexpression of mutant forms of LRRK2 is associated with neurite shortening but knockout neurons have longer neurites in vitro [59, 60]. Although there is an obvious limitation in that none of these phenotypes are neurodegenerative, the balance of evidence suggests that the mutations have the opposite effect as knockout.
If we add together the observations that mutant forms of LRRK2 are active in biochemical assays with the in vivo observations that mutant alleles and knockouts produce opposite phenotypes, then, we can infer that mutations in LRRK2 enhance function. Assuming that LRRK2 is also the gene in the GWAS-nominated locus for PD risk, this further implies that mutations enhance normal LRRK2 function, although a completely neomorphic function cannot be entirely ruled out.
At this time, my working model is that although mutations in different domains of LRRK2 have slightly different biochemical effects, they all enhance the normal function of LRRK2, leading to excessive or persistent activation. It is therefore important to know what LRRK2 is doing at a cellular level. Additionally, understanding how LRRK2 relates to other PD genes might provide important clues as to whether those functions are related to pathogenesis.
Relationship of the Function of LRRK2 to Other PD Genes
Having established some of the inherent biochemical activities of LRRK2, it is important to next understand what function(s) LRRK2 performs in cells. Two important clues to function come from localization and from interaction partners.
Overexpressed LRRK2 is excluded from the nucleus and largely cytosolic [18] although some studies suggested a mitochondrial pool of the protein [17]. At low levels of expression, and possibly also with endogenous protein, the cytosolic localization of LRRK2 can be refined. In cells, LRRK2 is associated with vesicular structures including areas of endocytic uptake at the plasma membrane as well as autophagic vesicles and multivesicular bodies [61]. Similar vesicular localization has been noted in the brain [61–63] although some caution might be needed as antibodies against LRRK2 can have a high background [64]. In some contexts, particularly with mutations or after inhibition of kinase activity, LRRK2 can be found in inclusions in the cell [18, 31]. These inclusions may be associated with microtubules [65], perhaps because LRRK2 can bind to tubulin heterodimers [66, 67]. As discussed above, the common biochemical event shared between mutant and kinase-inhibited LRRK2 is dephosphorylation and loss of 14-3-3 binding [31]. Therefore, LRRK2 associates with vesicular structures and with cytoskeletal elements in a regulated fashion. Some of the regulation occurs by phosphorylation, as is often seen in signaling pathways, and some of the localization relates to protein interaction partners.
These data also suggest that LRRK2 might have a role in vesicular dynamics, particularly autophagy. Supporting this idea, LRRK2 knockout mice and rats have prominent changes in autophagy markers and attendant morphological changes [68•, 69, 70]. Such changes are predominantly seen in tissues where LRRK2 is highly expressed, but the homologue LRRK1 is not present, such as kidney, suggesting a degree of overlap in function between the two genes. Interestingly, in knockout mice kidney, there are higher levels of LC3-II at young ages but lower levels at higher ages [68•]. This would suggest that LRRK2 knockout is associated initially with enhanced autophagy, i.e., that LRRK2 is normally a suppressor of autophagic flux. Consistent with this idea, acute LRRK2 kinase inhibition in cells causes enhanced autophagy in a LRRK2-dependent manner [71] and mutations in LRRK2 limit autophagy [72••]. The changes towards the opposite direction in older animals likely represent a compensatory pathway limiting the effects of loss of LRRK2 over time.
Vesicular sorting and transport in many organisms involves the interaction of proteins at the surface of lipid membranes, particularly members of the Rab family of small GTPases, and the cytoskeleton [73]. Therefore, the localization of LRRK2 to vesicles and cytoskeleton would indicate that it plays a role in vesicular sorting. Consistent with this idea, knockout of the single lrrk gene in Drosophila causes alterations in lysosomal position, a Rab-dependent phenomenon [74].
Collectively, these observations lead to the broader question of whether, outside of 14-3-3 and tubulin, additional protein interaction partners might influence LRRK2 localization and, hence, function. There are many proteins that have been claimed to interact with LRRK2 at least in some conditions. I do not have space to enumerate all of them here, but the interested reader is directed to some recent summary articles that have attempted to report systematically on the strength of evidence for each interaction [75, 76]. Instead, I will focus on a smaller number of examples that illustrate the larger point about localization being related to function, some coming from my own laboratory.
Several years ago, we started a project aimed at finding novel LRRK2 interacting proteins using an entirely in vitro strategy. We purified full-length, recombinant LRRK2 and LRRK1 [49] which we then used to probe commercially available (Invitrogen) glass microarrays containing ∼10,000 recombinant proteins generated in vitro. Such protoarrays have been used to identify protein interactions for several other mammalian proteins [77–79] and have the potential utility of having all target proteins expressed at the same level. Across several experiments [80, 81•], we recovered many known interactors of LRRK2, such as 14-3-3 proteins [30, 31], and LRRK1, such as EGFR [82, 83]. We also found BAG5, which had previously been nominated as a LRRK2 interactor by a yeast two-hybrid screen [84]. BAG proteins are known adaptors for Hsp70/Hsc70 [85] that in turn had previously been identified as a LRRK2 interactor [86]. Overall, these results show that protoarrays can be used to identify known, and hence valid, LRRK2 interaction partners.
More interestingly, we recovered two additional interactors that initially seemed to be unrelated to any known LRRK2 function, namely Rab7L1 and GAK. Rab7L1 is a small GTPase that had previously been shown to be localized to the Golgi network [87]. GAK and its homologue auxilin are well characterized to have roles in uncoating of clathrin-coated vesicles [88], with GAK having a specific role at the Golgi network [89]. We were able to show that LRRK2 binds Rab7L1 and GAK in a single complex that appeared to be stabilized by the BAG/HSc70 co-chaperone/chaperone pairing. In cellular experiments, we found that the overall complex promoted the turnover of a subset of vesicles derived from the trans-Golgi network (TGN) via a mechanism that involves the autophagy-lysosome pathway [81•]. Although this result needs further mechanistic exploration, particularly in understanding whether the same events occur at the endogenous level of LRRK2 expression, we have been able to validate these results using an automated system [32]. Interestingly, the Rab7L1-induced relocalization of LRRK2 to the TGN depends on expression of ArhGEF7, a proposed LRRK2 substrate [47]. This result raises the possibility that LRRK2 kinase activity may be important in vesicular sorting and turnover.
Independently, Rab7L was nominated as a LRRK2-interacting protein based on analysis of the PARK16 locus nominated by GWAS as containing a gene or genes associated with PD risk [90•]. Macleod et al. also proposed that LRRK2 interacts physically with VPS35, a component of the retromer complex that controls recycling of proteins from the endosomal system to the TGN [91]. Importantly, mutations in VPS35 cause an autosomal dominant form of PD; thus, the nominated interaction between LRRK2 and retromer unites two familial forms of PD [92, 93].
Although both studies concurred that LRRK2 and Rab7L1 interact physically, there are some unresolved discrepancies. For example, Macleod et al. proposed that increased expression of Rab7L1 rescues LRRK2-induced neurite shortening and, consistent with this, lower expression variants of Rab7L1 is associated with PD risk [90•]. In contrast, our analysis proposed that both increased expression of Rab7L1 and expression of any pathogenic mutant form of LRRK2 increased relocalization to the TGN, i.e., both proteins acted in the same direction [81•]. Additionally, in human brain, higher expression of Rab7L1 was associated with PD risk using two different RNA expression measurements [81•, 94••].
It is possible that Rab7L1 could promote LRRK2 relocalization to the TGN while also limiting neurite shortening as there is no need to assume a direct correlation between these two events. However, the human brain expression data should be able to be unambiguously resolved in the future. Expression quantitative trait loci (eQTL) can be identified by comparing gene variants with gene expression, usually measured using microarrays or RNA-seq [95]. Because the effects of risk variants on gene expression are often small, findings from a single series can be difficult to interpret without replication. It will therefore be important to confirm or refute the association between risk variants at the PARK16 locus and expression of Rab7L1 in additional brain series and to understand the direction of effect.
Although there are, therefore, some outstanding questions about Rab7L1, it is highly likely that this gene contributes to some of the risk at the PARK16 locus. GAK, which we also found interacting with LRRK2, is also a replicated risk locus for sporadic PD [13, 14, 94••]. Whether GAK explains all of the risk of PD is less certain than for Rab7L1 as the locus contains two independent signals that may indicate more than one gene contributes at this region. However, there are some genetic data that support GAK as a candidate for a PD gene. There are reports that loss of function mutations in the GAK homologue auxilin (gene name DNAJC6) cause early onset parkinsonism [96, 97]. Additionally, there are mutations in the auxilin interaction partner, synaptojanin-1 (SYNJ1), that also cause early onset parkinsonism [98, 99]. Collectively, these results suggest that the biological process of clathrin uncoating may be important in PD-related pathways.
Although there is work to be done in confirming that LRRK2 has multiple binding partners, they show that protein interactions tie together genes relevant for both inherited and sporadic PD. In other words, functional complexes of proteins working in defined cellular pathways explain at least aspects of the disease process. It is therefore of interest to know how many genes for PD we can explain by protein interactions. As discussed elsewhere [100], LRRK2 has strong links to at least two other important genes for parkinsonism, SNCA and MAPT, that code for neuronal proteins involved in endocytosis and cytoskeletal stability and likely related to the identified function of LRRK2 in vesicular sorting along cytoskeletal tracks.
Conclusions
The data available to date suggests that LRRK2 plays a critical role in the underlying pathobiological processes relevant to inherited and sporadic PD. Understanding the function of a protein complex that includes LRRK2 has allowed conceptual extension to several different genes causing PD. By extension, this suggests that further discoveries in the realm of PD genetics might be useful in development of therapeutics for multiple forms of disease.
At the same time, there are several areas where our understanding is not yet fully developed. Several of the individual results, including some published from my lab, require confirmation or refutation by others. Globally, whether all PD genes, including those that cause recessive parkinsonism, are related to the same set of pathways remains uncertain. Most importantly, how we get from genes to parkinsonism is uncertain as none of the nominated genes or processes are restricted to neurons that are sensitive to PD, such as dopaminergic neurons in the substantia nigra. However, that we can now acknowledge that there are additional areas that need to be explored shows how far our knowledge of the etiopathogenesis of PD has developed in the past few years. It is my distinct hope that this will continue over the next few years to the point where some of this work can be returned to people living with PD in the form of novel therapeutics.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Foley AR, Menezes MP, Pandraud A, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain. 2014;137:44–56. doi:10.1093/brain/awt315.
Amin R, Ratjen F. Emerging drugs for cystic fibrosis. Expert Opin Emerg Drugs. 2014;19:143–55. doi:10.1517/14728214.2014.882316.
Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7.
Paisán-Ruíz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi:10.1016/j.neuron.2004.10.023.
Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7. doi:10.1016/j.neuron.2004.11.005.
Funayama M, Hasegawa K, Ohta E, et al. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol. 2005;57:918–21. doi:10.1002/ana.20484.
Kay DM, Kramer P, Higgins D, et al. Escaping Parkinson’s disease: a neurologically healthy octogenarian with the LRRK2 G2019S mutation. Mov Disord. 2005;20:1077–8. doi:10.1002/mds.20618.
Bardien S, Lesage S, Brice A, Carr J. Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Parkinsonism Relat Disord. 2011;17:501–8. doi:10.1016/j.parkreldis.2010.11.008.
Benamer HTS, de Silva R. LRRK2 G2019S in the North African population: a review. Eur Neurol. 2010;63:321–5. doi:10.1159/000279653.
Troiano AR, Elbaz A, Lohmann E, et al. Low disease risk in relatives of North African lrrk2 Parkinson disease patients. Neurology. 2010;75:1118–9. doi:10.1212/WNL.0b013e3181f39a2e.
Simón-Sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12. doi:10.1038/ng.487.
Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–7. doi:10.1038/ng.485.
Do CB, Tung JY, Dorfman E, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011;7, e1002141. doi:10.1371/journal.pgen.1002141.
Lill CM, Roehr JT, McQueen MB, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genet. 2012;8, e1002548. doi:10.1371/journal.pgen.1002548.
Singleton A, Hardy J. A generalizable hypothesis for the genetic architecture of disease: pleomorphic risk loci. Hum Mol Genet. 2011;20:R158–62. doi:10.1093/hmg/ddr358.
Mata IF, Wedemeyer WJ, Farrer MJ, et al. LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–93. doi:10.1016/j.tins.2006.03.006.
West AB, Moore DJ, Biskup S, et al. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–7. doi:10.1073/pnas.0507360102.
Greggio E, Jain S, Kingsbury A, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–41. doi:10.1016/j.nbd.2006.04.001.
Greggio E, Cookson MR. Leucine-rich repeat kinase 2 mutations and Parkinson’s disease: three questions. ASN Neuro. 2009. doi:10.1042/AN20090007.
Smith WW, Pei Z, Jiang H, et al. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–3. doi:10.1038/nn1776.
Lee BD, Shin J-H, VanKampen J, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med. 2010;16:998–1000. doi:10.1038/nm.2199.
Deng X, Dzamko N, Prescott A, et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat Chem Biol. 2011;7:203–5. doi:10.1038/nchembio.538.
Göring S, Taymans J-M, Baekelandt V, Schmidt B. Indolinone based LRRK2 kinase inhibitors with a key hydrogen bond. Bioorg Med Chem Lett. 2014;24:4630–7. doi:10.1016/j.bmcl.2014.08.049.
Henderson JL, Kormos BL, Hayward MM, et al. Discovery and preclinical profiling of 3-[4-(Morpholin-4-yl)-7H-pyrrolo[2,3-d]pyrimidin-5-yl]benzonitrile (PF-06447475), a highly potent, selective, brain penetrant, and in vivo active LRRK2 kinase inhibitor. J Med Chem. 2014. doi:10.1021/jm5014055.
Sheng Z, Zhang S, Bustos D, et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med. 2012;4:164ra161. doi:10.1126/scitranslmed.3004485.
Yao C, Johnson WM, Gao Y, et al. Kinase inhibitors arrest neurodegeneration in cell and C. elegans models of LRRK2 toxicity. Hum Mol Genet. 2012. doi:10.1093/hmg/dds431.
Fuji RN, Flagella M, Baca M, et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci Transl Med. 2015;7:273ra15. doi:10.1126/scitranslmed.aaa3634.
Rudenko IN, Kaganovich A, Hauser DN, et al. The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem J. 2012;446:99–111. doi:10.1042/BJ20120637.
Rudenko IN, Chia R, Cookson MR. Is inhibition of kinase activity the only therapeutic strategy for LRRK2-associated Parkinson’s disease? BMC Med. 2012;10:20. doi:10.1186/1741-7015-10-20.
Li X, Wang QJ, Pan N, et al. Phosphorylation-dependent 14-3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson’s disease. PLoS ONE. 2011;6, e17153. doi:10.1371/journal.pone.0017153.
Nichols RJ, Dzamko N, Morrice NA, et al. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem J. 2010;430:393–404. doi:10.1042/BJ20100483.
Chia R, Haddock S, Beilina A, et al. Phosphorylation of LRRK2 by casein kinase 1α regulates trans-Golgi clustering via differential interaction with ARHGEF7. Nat Commun. 2014;5:5827. doi:10.1038/ncomms6827.
Dzamko N, Inesta-Vaquera F, Zhang J, et al. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS ONE. 2012;7, e39132. doi:10.1371/journal.pone.0039132.
Lobbestael E, Zhao J, Rudenko IN, et al. Identification of protein phosphatase 1 as a regulator of the LRRK2 phosphorylation cycle. Biochem J. 2013;456:119–28. doi:10.1042/BJ20121772.
Dzamko N, Deak M, Hentati F, et al. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J. 2010;430:405–13. doi:10.1042/BJ20100784.
Skibinski G, Nakamura K, Cookson MR, Finkbeiner S. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J Neurosci. 2014;34:418–33. doi:10.1523/JNEUROSCI.2712-13.2014.
Marín I, van Egmond WN, van Haastert PJM. The Roco protein family: a functional perspective. FASEB J. 2008;22:3103–10. doi:10.1096/fj.08-111310.
Gasper R, Meyer S, Gotthardt K, et al. It takes two to tango: regulation of G proteins by dimerization. Nat Rev Mol Cell Biol. 2009;10:423–9. doi:10.1038/nrm2689.
Guo L, Gandhi PN, Wang W, et al. The Parkinson’s disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007;313:3658–70. doi:10.1016/j.yexcr.2007.07.007.
Lewis PA, Greggio E, Beilina A, et al. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. 2007;357:668–71. doi:10.1016/j.bbrc.2007.04.006.
West AB, Moore DJ, Choi C, et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–32. doi:10.1093/hmg/ddl471.
Stafa K, Trancikova A, Webber PJ, et al. GTPase activity and neuronal toxicity of Parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012;8, e1002526. doi:10.1371/journal.pgen.1002526.
Liao J, Wu C-X, Burlak C, et al. Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc Natl Acad Sci U S A. 2014. doi:10.1073/pnas.1323285111.
Li X, Tan Y-C, Poulose S, et al. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J Neurochem. 2007;103:238–47. doi:10.1111/j.1471-4159.2007.04743.x.
Dzamko N, Zhou J, Huang Y, Halliday GM. Parkinson’s disease-implicated kinases in the brain; insights into disease pathogenesis. Front Mol Neurosci. 2014;7:57. doi:10.3389/fnmol.2014.00057.
Xiong Y, Yuan C, Chen R, et al. ArfGAP1 is a GTPase activating protein for LRRK2: reciprocal regulation of ArfGAP1 by LRRK2. J Neurosci. 2012;32:3877–86. doi:10.1523/JNEUROSCI.4566-11.2012.
Haebig K, Gloeckner CJ, Miralles MG, et al. ARHGEF7 (Beta-PIX) acts as guanine nucleotide exchange factor for leucine-rich repeat kinase 2. PLoS ONE. 2010;5, e13762. doi:10.1371/journal.pone.0013762.
Berger Z, Smith KA, Lavoie MJ. Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry. 2010;49:5511–23. doi:10.1021/bi100157u.
Civiero L, Vancraenenbroeck R, Belluzzi E, et al. Biochemical characterization of highly purified leucine-rich repeat kinases 1 and 2 demonstrates formation of homodimers. PLoS ONE. 2012;7, e43472. doi:10.1371/journal.pone.0043472.
Greggio E, Zambrano I, Kaganovich A, et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem. 2008;283:16906–14. doi:10.1074/jbc.M708718200.
Ito G, Iwatsubo T. Re-examination of the dimerization state of leucine-rich repeat kinase 2: predominance of the monomeric form. Biochem J. 2012;441:987–94. doi:10.1042/BJ20111215.
Gotthardt K, Weyand M, Kortholt A, et al. Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. EMBO J. 2008;27:2239–49. doi:10.1038/emboj.2008.150.
Korr D, Toschi L, Donner P, et al. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal. 2006;18:910–20. doi:10.1016/j.cellsig.2005.08.015.
Taymans J-M, Vancraenenbroeck R, Ollikainen P, et al. LRRK2 kinase activity is dependent on LRRK2 GTP binding capacity but independent of LRRK2 GTP binding. PLoS ONE. 2011;6, e23207. doi:10.1371/journal.pone.0023207.
Liu M, Kang S, Ray S, et al. Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry. 2011;50:9399–408. doi:10.1021/bi201173d.
Ray S, Bender S, Kang S, et al. The Parkinson disease-linked LRRK2 protein mutation I2020T stabilizes an active state conformation leading to increased kinase activity. J Biol Chem. 2014;289:13042–53. doi:10.1074/jbc.M113.537811.
Sen S, Webber PJ, West AB. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J Biol Chem. 2009;284:36346–56. doi:10.1074/jbc.M109.025437.
Herzig MC, Kolly C, Persohn E, et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20:4209–23. doi:10.1093/hmg/ddr348.
MacLeod D, Dowman J, Hammond R, et al. The familial parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–93. doi:10.1016/j.neuron.2006.10.008.
Dächsel JC, Behrouz B, Yue M, et al. A comparative study of Lrrk2 function in primary neuronal cultures. Parkinsonism Relat Disord. 2010;16:650–5. doi:10.1016/j.parkreldis.2010.08.018.
Alegre-Abarrategui J, Christian H, Lufino MMP, et al. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet. 2009;18:4022–34. doi:10.1093/hmg/ddp346.
Biskup S, Moore DJ, Celsi F, et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–69. doi:10.1002/ana.21019.
Higashi S, Biskup S, West AB, et al. Localization of Parkinson’s disease-associated LRRK2 in normal and pathological human brain. Brain Res. 2007;1155:208–19. doi:10.1016/j.brainres.2007.04.034.
Davies P, Hinkle KM, Sukar NN, et al. Comprehensive characterization and optimization of leucine rich repeat kinase 2 (LRRK2) monoclonal antibodies. Biochem J. 2013. doi:10.1042/BJ20121742.
Kett LR, Boassa D, Ho CC-Y, et al. LRRK2 Parkinson disease mutations enhance its microtubule association. Hum Mol Genet. 2012;21:890–9. doi:10.1093/hmg/ddr526.
Law BMH, Spain VA, Leinster VHL, et al. A direct interaction between leucine-rich repeat kinase 2 and specific β-tubulin isoforms regulates tubulin acetylation. J Biol Chem. 2014;289:895–908. doi:10.1074/jbc.M113.507913.
Caesar M, Zach S, Carlson CB, et al. Leucine-rich repeat kinase 2 functionally interacts with microtubules and kinase-dependently modulates cell migration. Neurobiol Dis. 2013;54:280–8. doi:10.1016/j.nbd.2012.12.019.
Tong Y, Giaime E, Yamaguchi H, et al. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol Neurodegener. 2012;7:2. doi:10.1186/1750-1326-7-2. This paper shows how phenotypes in LRRK2 knockout mice are affected by age. The exact interpretation of this data is not yet clear, but likely indicate that phenotypes are a mix of direct effect of loss of LRRK2 and compensatory changes in the same pathway.
Tong Y, Yamaguchi H, Giaime E, et al. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107:9879–84. doi:10.1073/pnas.1004676107.
Baptista MAS, Dave KD, Frasier MA, et al. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS ONE. 2013;8, e80705. doi:10.1371/journal.pone.0080705.
Manzoni C, Mamais A, Dihanich S, et al. Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim Biophys Acta. 2013;1833:2900–10. doi:10.1016/j.bbamcr.2013.07.020.
Manzoni C, Mamais A, Dihanich S, et al. Pathogenic Parkinson’s disease mutations across the functional domains of LRRK2 alter the autophagic/lysosomal response to starvation. Biochem Biophys Res Commun. 2013;441:862–6. doi:10.1016/j.bbrc.2013.10.159. Along with companion paper 71, the study by Manzoni et al is one of the first to show consistent biochemical effects of LRRK2 mutations in an autophagy related pathway. Of interest, the direction of effect of mutations is opposite that of kinase inhibitors, supporting the idea that mutations have a gain of normal function.
Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–17. doi:10.1038/35052055.
Dodson MW, Zhang T, Jiang C, et al. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet. 2012;21:1350–63. doi:10.1093/hmg/ddr573.
Manzoni C, Denny P, Lovering R, Lewis PA. Computational analysis of the LRRK2 interactome. PeerJ. 2015;3, e778.
Porras P, Duesbury M, Fabregat A, et al. A visual review of the interactome of LRRK2: using deep-curated molecular interactions data to represent biology. Proteomics. 2015. doi:10.1002/pmic.201400390.
Fenner BJ, Scannell M, Prehn JHM. Expanding the substantial interactome of NEMO using protein microarrays. PLoS ONE. 2010;5, e8799. doi:10.1371/journal.pone.0008799.
Al-Mulla F, Bitar MS, Al-Maghrebi M, et al. Raf kinase inhibitor protein RKIP enhances signaling by glycogen synthase kinase-3β. Cancer Res. 2011;71:1334–43. doi:10.1158/0008-5472.CAN-10-3102.
Tong Y, Ben-Shlomo A, Zhou C, et al. Pituitary tumor transforming gene 1 regulates Aurora kinase A activity. Oncogene. 2008;27:6385–95. doi:10.1038/onc.2008.234.
Reyniers L, Del Giudice MG, Civiero L, et al. Differential protein-protein interactions of LRRK1 and LRRK2 indicate roles in distinct cellular signaling pathways. J Neurochem. 2014. doi:10.1111/jnc.12798.
Beilina A, Rudenko IN, Kaganovich A, et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A. 2014. doi:10.1073/pnas.1318306111. This paper, which is from my laboratory in collaboration with several other groups, indicates a potential relationship between LRRK2 and two proteins in GWAS-nominated regions for sporadic PD risk. Reference 90 also shows an interaction between LRRK2 and Rab7L1.
Hanafusa H, Ishikawa K, Kedashiro S, et al. Leucine-rich repeat kinase LRRK1 regulates endosomal trafficking of the EGF receptor. Nat Commun. 2011;2:158. doi:10.1038/ncomms1161.
Ishikawa K, Nara A, Matsumoto K, Hanafusa H. EGFR-dependent phosphorylation of leucine-rich repeat kinase LRRK1 is important for proper endosomal trafficking of EGFR. Mol Biol Cell. 2012;23:1294–306. doi:10.1091/mbc.E11-09-0780.
Zheng X, Yang M, Tan J, et al. Screening of LRRK2 interactants by yeast 2-hybrid analysis. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2008;33:883–91.
Kabbage M, Dickman MB. The BAG proteins: a ubiquitous family of chaperone regulators. Cell Mol Life Sci. 2008;65:1390–402. doi:10.1007/s00018-008-7535-2.
Dächsel JC, Taylor JP, Mok SS, et al. Identification of potential protein interactors of Lrrk2. Parkinsonism Relat Disord. 2007;13:382–5. doi:10.1016/j.parkreldis.2007.01.008.
Helip-Wooley A, Thoene JG. Sucrose-induced vacuolation results in increased expression of cholesterol biosynthesis and lysosomal genes. Exp Cell Res. 2004;292:89–100.
Eisenberg E, Greene LE. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic. 2007;8:640–6. doi:10.1111/j.1600-0854.2007.00568.x.
Zhang CX, Engqvist-Goldstein AEY, Carreno S, et al. Multiple roles for cyclin G-associated kinase in clathrin-mediated sorting events. Traffic. 2005;6:1103–13. doi:10.1111/j.1600-0854.2005.00346.x.
Macleod DA, Rhinn H, Kuwahara T, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77:425–39. doi:10.1016/j.neuron.2012.11.033. This paper, along with reference 81, showed that LRRK2 interacts with the GWAS candidate gene Rab7L1. In this paper, the authors also indicate an effect on VPS35, part of the retromer complex and a gene for inherited PD (see references 92 and 93).
Cullen PJ, Korswagen HC. Sorting nexins provide diversity for retromer-dependent trafficking events. Nat Cell Biol. 2012;14:29–37. doi:10.1038/ncb2374.
Vilariño-Güell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–7. doi:10.1016/j.ajhg.2011.06.001.
Zimprich A, Benet-Pagès A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–75. doi:10.1016/j.ajhg.2011.06.008.
Nalls MA, Pankratz N, Lill CM, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46:989–93. doi:10.1038/ng.3043. This is the latest iteration of GWAS in PD, using data combined from many groups around the world and indicating that sporadic PD risk is influenced by more than twenty independent genetic factors.
Pandey AK, Williams RW. Genetics of gene expression in CNS. Int Rev Neurobiol. 2014;116:195–231. doi:10.1016/B978-0-12-801105-8.00008-4.
Köroğlu Ç, Baysal L, Cetinkaya M, et al. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord. 2013;19:320–4. doi:10.1016/j.parkreldis.2012.11.006.
Edvardson S, Cinnamon Y, Ta-Shma A, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE. 2012;7, e36458. doi:10.1371/journal.pone.0036458.
Quadri M, Fang M, Picillo M, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset parkinsonism. Hum Mutat. 2013;34:1208–15. doi:10.1002/humu.22373.
Krebs CE, Karkheiran S, Powell JC, et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive parkinsonism with generalized seizures. Hum Mutat. 2013;34:1200–7. doi:10.1002/humu.22372.
Taymans J-M, Cookson MR. Mechanisms in dominant parkinsonism: the toxic triangle of LRRK2, alpha-synuclein, and tau. Bioessays. 2010;32:227–35. doi:10.1002/bies.200900163.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Aging. For reasons of space, I unfortunately was not able to quote all of the primary literature and apologize to those colleagues whose work is mentioned in other reviews and not included directly here.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Mark R. Cookson declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Genetics
Rights and permissions
About this article

Cite this article
Cookson, M.R. LRRK2 Pathways Leading to Neurodegeneration. Curr Neurol Neurosci Rep 15, 42 (2015). https://doi.org/10.1007/s11910-015-0564-y
Published:
DOI: https://doi.org/10.1007/s11910-015-0564-y