
Abstract
Disorders of glycogen metabolism are inborn errors of energy homeostasis affecting primarily skeletal muscle, heart, liver, and, less frequently, the central nervous system. These rare diseases are quite variable in age of onset, symptoms, morbidity, and mortality. This review provides an update on disorders of glycogen metabolism affecting skeletal muscle exclusively or predominantly. From a pathogenetic perspective, we classify these diseases as primary, if the defective enzyme is directly involved in glycogen/glucose metabolism, or secondary, if the genetic mutation affects proteins which indirectly regulate glycogen or glucose processing. In addition to summarizing the most recent clinical reports in this field, we briefly describe animal models of human glycogen disorders. These experimental models are greatly improving the understanding of the pathogenetic mechanisms underlying the muscle degenerative process associated to these diseases and provide in vivo platforms to test new therapeutic strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycogen is a highly branched polymer of glucose molecules connected by α-1,4 glycosidic linkages and branches of glucose molecules connected to the chains from which they branch off by α-1,6 glycosidic bonds between the first glucose molecule of the new branch and a glucose molecule in the stem. The presence of dispersed glycogen granules in cells, and the accompanying enzymes, ensures a rapidly accessible source of cellular energy [1]. The defects of muscle glycogen metabolism (glycogenoses or glycogen storage diseases), classically numbered according to the specific enzyme defect and chronology of their discovery, can cause (1) pathological accumulation of normal-structure glycogen molecules, (2) pathological accumulation of abnormal-structure glycogen molecules, and (3) glycogen depletion.
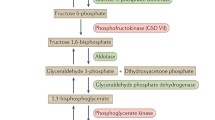
Clinically, these disorders result in two main syndromes: exercise intolerance, with cramps and myoglobinuria, and progressive weakness. In recent years, new clinical entities have been described and novel physiopathological aspects have been clarified [2–4]. Accordingly, we suggest a novel physiopathological classification which further distinguishes between primary and secondary glycogenoses (Table 1). We will define primary muscle glycogen diseases as those caused by genetic defects in the enzymes directly involved in glycogen synthesis (gluconeogenesis), glycogen degradation (glycogenolysis), or glucose metabolism (glycolysis), and secondary glycogenoses as those caused by the functional loss of regulatory proteins that may indirectly affect the enzymes specifically active in glycogen or glucose pathways (Fig. 1).

Primary and secondary glycogenoses can lead to (1) pathological accumulation of normal-structure glycogen molecules, (2) pathological accumulation of abnormal-structure glycogen molecules, and (3) glycogen depletion. In primary forms, genetic defects affect enzymes directly involved in glycogen synthesis or lysis. In the secondary disorders, genetic mutations concern molecules which indirectly regulate glycogen or glucose metabolism by modulating the action of the primary enzymes. APBD adult polyglucosan body disease, FCNLHG Fatal congenital nonlysosomal heart glycogenosis, FHC familial hypertrophic cardiomyopathy, GSD glycogen storage disease, PhK phosphorylase kinase, WPWS Wolff–Parkinson–White syndrome
This article focuses on the recent physiopathological and clinical data published in this field and on the murine models which have been generated for these disorders (Table 2). Although glycogenosis type II (Pompe disease) is frequent [5], the complexity of this disorder requires a specific description in a dedicated review and hence it will not be addressed in this article.
Primary Glycogenoses
Defects of Glycogen Synthesis
Biosynthesis of glycogen is accomplished in three main steps. First, the glycosyltransferase glycogenin-1, encoded by the gene GYG1, catalyzes the formation of a short glucose polymer, of approximately ten glucose residues, from uridine diphosphate glucose through an autoglucosylation reaction. This short oligosaccharide is then extended by glycogen synthase (GS), encoded by GYS1 in muscle, until a 13-mer glycogenin-bound glucose polymer is produced. Then, glycogen branching enzyme (GBE) detaches the last six glucoses as a hexamer, reattaching it to the side of the remaining heptamer, converting the original linear polymer to a fork with two ends. Finally, GS extends each end, and GBE removes each new hexamer and causes it to branch as described above, continuing the process until the tightly packed glycogen sphere is formed [4].
Glycogenoses Type 0 and Type XV (“Aglycogenoses”)
Glycogen depletion was first identified in an adult patient with cardiomyopathy and muscle weakness associated with muscle glycogen depletion. The patient was affected by muscle glycogenin-1 deficiency. In the GYG1 gene, a missense mutation on one allele (p.T83M) and a nonsense mutation on the other (p.A163TfsX5) were identified [6, 7].
A second defect of glycogen synthesis was described in three siblings who displayed cardiomyopathy and skeletal myopathy. These patients carried a homozygous c.1384C > T transition in exon 11 of GYS1 resulting in a truncated muscle GS protein. The clinical manifestations started in childhood and included exercise intolerance, increased cardiac mass, cardiac arrhythmias, and normal glucose tolerance test findings [8]. In 2003, Pederson et al. [9] generated a GS knockout mouse model. About 90 % of the homozygous null mice die perinatally due to abnormal cardiac development and congenital heart disease is one of the commonest birth defects in newborn mice. In both GS and glycogenin-1 defects, muscle biopsy shows absence of muscle glycogen and predominance of oxidative fibers with marked mitochondrial accumulation.
Glycogenosis Type IV
The disease is characterized by accumulation of an abnormal glycogen with fewer branching points, more 1,4-linked glucose units and longer outer chains [an amylopectin-like structure or polyglucosan (PG)] than normal. PG bodies are periodic acid–Schiff (PAS)-positive, diastase-resistant, and accumulate in tissues with high metabolic activity such as liver, skeletal muscles, amniocytes, and the central nervous system [10]. To date, 38 mutations in the gene encoding GBE (GBE1) have been identified in patients affected by glycogenosis type IV [11, 12]. The severity of phenotypes associated with GBE deficiency appears to correlate with the “molecular severity” of the mutations in GBE1: perinatal death has been invariably associated with severe mutations, whereas milder, nonlethal neuromuscular cases had more than 5 % residual GBE activity in fibroblasts due to milder missense mutations [13].
Neuromuscular presentation of glycogenosis type IV is heterogeneous with four main variants based on the amount of residual enzyme activity. The perinatal form (meaning both prenatal and postnatal) presents as fetal akinesia deformation sequence and is characterized by multiple congenital contractures (arthrogryposis multiplex congenita), hydrops fetalis, and perinatal death. The second form, labeled rather generically as “congenital” or “fatal infantile,” presents at or soon after birth with hypotonia, muscle wasting, neuronal involvement, inconsistent cardiomyopathy, and early death. The juvenile phenotype is dominated by either myopathy [13, 14] or cardiopathy [15], whereas the adult form can present as isolated myopathy [16] or as a multisystem disorder with central and peripheral nervous system dysfunction (adult PG body disease) [17, 18].
The diagnosis of glycogenosis type IV can be made by histologic and ultrastructural examination [10], and can be confirmed by the determination of GBE activity in affected tissues. Muscle biopsies show cytoplasmic vacuoles containing PAS-positive and diastase-resistant glycogen material. Electron-microscopic examination displays finely granular, electron-lucent vacuoles filled with amylopectin-like material identified as PG [10, 19].
Although two naturally occurring glycogenosis type IV animal models have been described [20, 21], two independent mouse models of glycogenosis type IV have been generated. Lee et al. [22] inserted a stop codon mutation (p.E609X) in the Gbe1 gene using a gene-driven mutagenesis approach. Homozygous mutants (Gbe −/−) recapitulate the clinical features of hydrops fetalis and the embryonic lethality of the severe fetal form of glycogenosis type IV. Little PG deposition is detected in the embryonal heart, liver, and skeletal muscle; no cell injury is observed. Histologically, the mutant mice display hyperproliferation of cardiomyocytes and hypertrabeculation/noncompaction of the ventricular wall, suggesting that cardiac glycogen might play a critical role in regulating normal heart development and maturation [22]. Akman et al. [23] created two additional mouse models of GBE deficiency. In the first one, they decreased GBE expression via transcriptional interference by a reverse-oriented phosphoglycerate kinase (PGK)–neomycin cassette. The construct leads to a hypomorphic allele with residual enzyme activity and later onset of disease, yet a pronounced accumulation of PG in the brain, heart, skeletal muscle, and liver. In the second model, mutant mice carry a Gbe1 exon 7 deletion, which completely abolishes GBE synthesis. The mice die at or soon after birth and show PG accumulation in the heart, liver, and skeletal muscle. Although Akman et al. [23] did not observe the hydropic embryos or left ventricular noncompaction noted in patients, the hearts of Gbe1 −/− mice have disrupted myocardial architecture in association with widespread PG accumulation [23].
Defects of Glycogenolysis
Glycogenolysis is stimulated by phosphorylase b kinase (PHK), which, in response to various neural and hormonal signals, phosphorylates and activates muscle glycogen phosphorylase or myophosphorylase. The enzyme removes 1,4-glycosyl residues from the glycogen molecule, liberating glucose 1-phosphate, which can enter the glycolytic pathway. After phosphorylase has shortened the peripheral chains of glycogen to about four glycosyl units, these residual tail ends are removed by glycogen debranching enzyme (GDE) in two steps catalyzed by an oligo-1,4-1,4-glucantransferase and by an amylo-1,6-glucosidase [1].
Glycogenosis Type VIII
PHK is a multimeric enzyme composed of four different subunits, α, β, γ, and δ. The γ subunit is catalytic and is regulated by the degree of phosphorylation of the α and β subunits. Calcium sensitivity is conferred by the δ subunit, which is tightly bound to calmodulin [1]. PHK deficiency has been associated with five syndromes distinguished by inheritance and by tissue involvement [24•]: (1) a benign X-linked recessive hepatopathy of infancy or childhood; (2) an autosomal recessive liver and muscle disease; (3) a pure myopathy with a muscle biopsy showing a modest accumulation of glycogen; (4) an autosomal recessive severe liver disease with cirrhosis; and (5) a fetal infantile cardiopathy, reported in a handful of patients.
The pure myopathy is due to mutations in the X-linked gene (PhKA1) encoding the muscle-specific α subunit [24•]. Muscle PHK deficiency ranges from exercise intolerance resembling defects of myophosphorylase to an almost asymptomatic condition [25–29]. This milder phenotype is probably explained by redundant myophosphorylase activation pathways, which bypass PHK defects [30, 31].
Glycogenosis Type V (McArdle Disease)
Glycogenosis type V is caused by deficiency of the muscle glycogen phosphorylase, also called myophosphorylase. The disease is characterized by exercise intolerance triggered by activities involving isometric or dynamic exercise. Indeed, blockage of glycogen breakdown impairs the release of glucose and forces the muscle to rely on fatty acid oxidation and uptake of glucose from the blood [32]. Myophosphorylase is encoded by the gene PYGM, which consists of 20 exons. The most prevalent PYGM mutation in the Caucasian population is a nonsense mutation located in exon 1 (p.R50X) resulting in nonsense-mediated decay of the PYGM messenger RNA [33, 34]. The second commonest mutations in the Caucasian population are the missense mutation p.G205S in exon 5 and the missense mutation p.W798R, which has been reported only in patients of Spanish origin [33].
Beyond these common mutants, the disease is characterized by a high genetic heterogeneity as substantiated by the documentation of more than 133 mutations to date [33, 35]. For most mutations it is not possible to identify a genotype/phenotype correlation. The only exception described so far is represented by two patients with atypical glycogenosis type V carrying the common mutations on one allele (R50X and G205S), and splice mutations in intron 3 [IVS3-26A4G (c.425-26A4G)] and intron 5 [IVS5-601G4A (c.856-601G4A)] on the other allele. These patients displayed a residual amount of enzyme and an atypical milder form of disease, thus suggesting that a low level of myophosphorylase could be sufficient to reduce the impact of the disease [36].
Although the clinical symptoms of glycogenosis type V begin during childhood and adolescence, diagnosis is rarely performed in young patients and is established mainly in patients in early adulthood (20–30 years of age) [37]. The four cardinal features of the disease are (1) exercise intolerance, i.e., reversible, acute crises of early fatigue and contractures, sometimes with rhabdomyolysis and myoglobinuria, triggered by static muscle contractions (lifting weights) or dynamic exercise (climbing stairs, running); (2) high serum creatine kinase (CK) levels in the absence of heavy exercise; (3) at least one episode of exertional hyperCKemia (i.e., at least several thousand units per liter) after intense exercise, indicating marked rhabdomyolysis frequently accompanied by myoglobinuria; (4) the “second wind” phenomenon, which is pathognomonic for glycogenosis type V in adults [38]. It can be defined as a decrease in perceived exertion and heart rate despite no change in the work rate. It occurs with or without a rest period and results from exercise-induced enhancement of muscle glycolysis made possible by increases of muscle blood flow and uptake of glucose from the blood.
For the diagnosis of glycogenosis type V, in any patient reporting acute crises of exercise intolerance accompanied by basal hyperCKemia, ideally an exercise confirmatory test should first be performed to objectively assess the “pathognomonic” second wind phenomenon. A second necessary step is PYGM genotyping, searching for the most prevalent mutations, i.e., mainly p.R50X, p.W798R, and p.G205S, which account, alone or in combination, for 61 % of all PYGM genotypes. Sequencing of all the coding regions of the gene should be performed when the search for the most prevalent mutations is unsuccessful and the muscle biopsy shows subsarcolemmal vacuolation, intermyofibrillar glycogen accumulation on PAS staining and electron microscopy and myophosphorylase deficiency on histochemistry [33, 39•].
Although two spontaneous animal models for glycogenosis type V have been identified, i.e., in Charolais cattle [40] and Merino sheep [41], they have rendered a limited amount of information on the pathophysiology of the disorder. Nogales-Gadea et al. [42] recently generated a knock-in mouse carrying the common p.R50X mutation. The mutant mice show absence of glycogen phosphorylase activity in muscle and subsequent blocked glycogenolysis, which results in a marked exercise intolerance and hyperCKemia that closely resemble the clinical presentation of the human disease .
Glycogenosis Type III
Glycogenosis type III is caused by deficiency of amylo-1,6- glucosidase, or GDE. Glycogenolysis is halted at the outermost branch points and accumulation of abnormal glycogen occurs in affected organs (liver, heart, skeletal muscle; leukocytes in glycogenosis type IIIa; liver in glycogenosis type IIIb) [43•]. Over 100 different mutations of the human GDE gene, AGL, have been reported to date in glycogenosis type IIIa patients and no genotype/phenotype correlation has been reported. In contrast, two mutations in exon 3, c.18_19delGA (p.Gln6HisfsX20) and c.16C > T (p.Gln6X), are specifically associated with glycogenosis type IIIb phenotype [44]. The clinical manifestations of GDE deficiency include hepatomegaly, hypoglycemia, short stature, dyslipidemia, progressive myopathy, and cardiomyopathy. Although hepatomegaly and hepatic symptoms in most individuals with glycogenosis type III improve with age and usually resolve after puberty, muscle weakness tends to be minimal during childhood, but may become the predominant feature during adulthood [43•]. Back pain and fatigue may be present, and exercise-induced myoglobinuria is common beyond childhood. Many patients show elevated serum CK levels. Neuropathy may occur owing to glycogen storage in Schwann cells and axons.
Glycogenosis type IIIa is associated with glycogen deposition in the myocardium. A subset of patients develop left ventricular hypertrophy, although on a histological basis, there is no myocyte disarray as seen in hypertrophic cardiomyopathy [45]. No correlation between the specific type of AGL mutation and left ventricular wall thickness or left ventricular mass exists. Regular screening of patients with glycogenosis type III with echocardiograms, including measurements of ventricular thickness as well as left ventricular mass and both systolic and diastolic function is strongly indicated. Evaluation of heart rhythm by electrocardiogram, Holter monitoring, or both is also reasonable as a screening test, given reports of GDE patients dying suddenly.
The diagnosis of glycogenosis type IIIa may be made by finding abnormal glycogen (limited dextrin with short outer branches) in liver and/or skeletal muscle biopsies. Biochemical tests with glycogen quantitation and determination of amylo-1,6- glucosidase activity in a liver/muscle biopsy specimen or erythrocytes can confirm the diagnosis.
As a result of the high level of genetic heterogeneity, full gene sequencing is typically required to identify mutations in the AGL gene. If glycogenosis type IIIb is suspected, exon 3 may be sequenced first, since exon 3 mutations are common causes of glycogenosis type IIIb. Genetic testing may obviate the need for more comprehensive and expensive sequencing or a muscle biopsy for enzyme analysis. If the individual has muscle involvement, screening for ethnic-group-specific mutations may be appropriate. If targeted mutation analysis fails to confirm the diagnosis, full AGL gene sequencing should be performed.
A glycogenosis type IIIa phenotype was identified in curly-coated retrievers, which carry a frameshift mutation leading to a deletion of the C-terminal 126 amino acids of GDE. Abnormally high glycogen deposition was observed in liver and muscle, and, gradually increasing activity of enzymes, including aspartate aminotransferase, alanine transferase, alkaline phosphatase, and CK, was found in serum. In muscle, increased glycogen deposition was accompanied by disruption of the contractile apparatus and fraying of myofibrils [46, 47].
Defects of Glycolysis
Glycogenosis Type VII
This disease is caused by deficiency of muscle phosphofructokinase (PFK), a key regulatory enzyme in glycolysis (ATP:d-fructose 6-phosphate-1-phosphotransferase) which catalyzes the irreversible conversion of fructose 6-phosphate to fructose 1,6-bisphosphate. PFK is a tetrameric enzyme derived from three distinct genetic loci, coding for muscle, liver, and platelet isoforms. Muscle and liver PFK are homotetramers of 4 M and 4 L subunits, respectively, whereas erythrocytes contain both L and M subunits [48].
When muscle PFK is deficient, glycolysis is affected not only in muscle, but also in erythrocytes, which typically show 50 % activity relative to normal. As erythrocytes have no mitochondria, this relatively mild reduction of glycolytic flux can critically reduce energy supply, resulting in hemolysis.
Muscle PFK deficiency causes four possible presentations of disease: (1) the classic form with exercise intolerance, myalgias, and myoglobinuria, (2) a severe infantile form, with hypotonia, developmental delay, dysmorphic features, corneal ulcers, progressive myopathy, cardiopathy, and respiratory failure, leading to early death in childhood, (3) a late-onset form, with “fixed” proximal myopathy, usually appearing in the fifth decade, and (4) a hemolytic form, characterized by nonspherocytic hemolytic anemia without muscle symptoms [49]. Clinical symptoms are more severe than in glycogenosis type V: they are usually already present during childhood, not relieved by the second wind phenomenon, and often accompanied by nausea and vomiting as well as compensated hemolytic anemia and hyperuricemia [50].
The diagnosis of glycogenosis type VII is based on muscle biopsy, which shows glycogen accumulation associated with histochemical PFK deficiency. Glycogen can often accumulate in PG bodies. It is feasible that the excessive accumulation of glucose 6-phosphate upstream of the glycolytic block activates GS, thus tilting the finely balanced activity ratio of GS and GBE in favor of GS and resulting in a polysaccharide with abnormally long and poorly branched chains [2]. Mutation analysis can provide further confirmation. About 20 mutations have been described in the PFKM gene [51], and no clear genotype/phenotype correlations have emerged.
García et al. [52] generated a knockout mouse model by deleting the 5′ promoter region and exon 3, which contains the translation start codon of the murine Pfkm gene. Pfkm-null mice show high early lethality and only few survive until adulthood. As in human patients, these mice show severe exercise intolerance and hemolysis. Lack of glycolysis in skeletal muscle also causes alterations in bioenergetics and compensatory changes in key metabolic genes. Limb and respiratory muscles present subsarcolemmal and intermyofibrillar glycogen accumulation with fiber degeneration and/or necrosis associated with multiple sites of regeneration. Moreover, the marked metabolic alterations in the heart lead to chronic hypertrophy.
Glycogenosis Type IX
Phosphoglycerate kinase (PGK) is encoded by the gene PGK1, which is ubiquitously expressed. The clinical presentations due to its deficiency depend on the isolated or combined involvement of three tissues: erythrocytes (hemolytic anemia), skeletal muscle (exercise intolerance, cramps, myoglobinuria), and the central nervous system (seizures, mental retardation, stroke) [53]. Muscle biopsy often revealed nonspecific changes.
In a review of 33 patients affected by PGK defects, Spiegel et al. [54] found that the commonest association was hemolytic anemia and central nervous system involvement (37 % of patients) . Interestingly, two unrelated patients carrying the p.T378P PGK1 mutation displayed the association of myopathy and severe juvenile Parkinsonism, suggesting that PGK defects should be considered in the diagnosis of this neurodegenerative disorder even when muscular symptoms are not yet present [54, 55].
Glycogenosis Type X
Phosphoglycerate mutase (PGAM) deficiency (glycogenosis type X) is a rare disorder caused by a partial block of terminal glycolysis [56]. PGAM is a dimeric enzyme composed of muscle-specific and brain-specific subunits. Mature human skeletal muscle contains the muscle-specific subunit and the brain-specific subunit, which accounts for the residual activity (usually less than 7 %) observed in PGAM-deficient patients.
In contrast to the homogeneous clinical phenotype, with onset in adolescence of exercise-induced cramps and myalgia often leading to recurrent myoglobinuria, DNA analysis revealed molecular heterogeneity. The commonest defect is a nonsense mutation at codon 78 (p.W78X) documented predominantly in African-American patients, either in homozygosity or in heterozygosity. In a few cases, muscle biopsy reveals diffuse or patchy glycogen accumulation, and in some patients tubular aggregates have been identified [56].
Glycogenosis Type XI
Lactate dehydrogenase (LDH) is a tetrameric enzyme composed of two subunits, M and H, resulting in five isozymes. LDH-M predominates in muscle and LDH-H in heart and other tissues. Muscle LDH deficiency (glycogenosis type XI) has been described in Japanese and Caucasian patients with exercise intolerance, myalgia, and myoglobinuria. In some cases, a characteristic skin rash has been observed. Muscle biopsy shows nonspecific myopathic changes and biochemical analysis reveals deficit of LDH activity. Molecular analysis of the LDH-M gene identified different mutations in these patients [57].
Glycogenosis Type XII
Aldolase A is responsible for the conversion of fructose 1,6-bisphosphate into glyceraldehyde 3-phosphate and dihydroxyacetone phosphate in the glycolytic pathway. Deficiency of aldolase A has been reported in a boy with exercise intolerance, muscle weakness, and hyperCKemia. Molecular analysis revealed the substitution of a single amino acid within the subunit interface, which is essential for the tetrameric structure of this enzyme [58].
Glycogenosis Type XIII
β-Enolase deficiency has been described in a patient with adult-onset easy muscle fatigability, myalgias, and increased CK levels after intense physical exertion. Muscle biopsy showed a residual enolase activity of about 5 %. The patient was heterozygous for two missense mutations in ENO3, which encodes the muscle isoform of β-enolase [59].
Glycogenosis Type XIV
A deficiency of phosphoglucomutase, the enzyme that catalyzes the conversion of glucose 1-phosphate to glucose 6-phosphate, was identified in a 35-year-old patient who presented with exercise intolerance with episodes of rhabdomyolysis. Muscle biopsy showed subsarcolemmal and sarcoplasmic glycogen accumulation, and muscle phosphoglucomutase activity was reduced to 1 % of the level in controls. The patient was compound heterozygous for two different missense mutations [60••].
Secondary Glycogenoses
Secondary glycogenoses are caused by genetic defects of proteins not directly involved in glycogen or glucose metabolism, but whose deficiency ultimately leads to the pathological accumulation of normal or abnormal glycogen molecules. In these disorders, skeletal muscle involvement is often overshadowed by the marked central nervous system or heart damage. Lafora disease and AMP-activated protein kinase (AMPK) deficiency are emblematic of this category of glycogenoses. However, alteration of muscle glycogen can be present also in autophagy vacuolar defects (i.e., Danon disease).
Lafora Disease
Lafora disease is the commonest cause of teenage-onset progressive myoclonus epilepsy. This disorder is fatal and is caused in large part by widespread progressive accumulation of PG masses of glycogen, also called Lafora bodies (LBs) [61]. Unlike GBE deficiency, in which PG bodies accumulate in axons/axon hillocks and produce an axonopathy, LBs aggregate in the somadendritic compartment and result in myoclonus epilepsy.
Seventy percent of Lafora disease patients display mutations in EPM2A, which encodes the protein laforin. This enzyme removes phosphate molecules erroneously incorporated on glycogen by GS [62], preventing the formation of phosphorylated, poorly branched insoluble glycogen polymers. Another function of laforin is to interact with malin/E3 ubiquitin ligase, the gene product of EPM2B, the second disease-causing gene, responsible for approximately 27 % of Lafora disease. Laforin and malin in concert cause proteasome-dependent degradation of protein-targeting glycogen (PTG), a molecule which activates GS. Through PTG degradation, laforin–malin ensures a blockade of neuronal glycogen synthesis under normal conditions. Notably, malin also regulates the polyubiquitination of laforin.
The pathogenesis of Lafora disease is complex and involves multiple levels of regulation. According to one hypothesis, absence of laforin or malin is associated with increased PTG level, most likely through loss of malin-mediated ubiquitination and degradation. This results in increased GS activity [63] and an imbalance between the actions of GS and GBE with consequent PG formation. According to a second hypothesis, when laforin is absent, phosphates accumulate on glycogen, disturbing its spherical structure and causing glycogen precipitation. On the other hand, the function of malin is (1) to prevent indiscriminate binding of laforin to glycogen by controlling its cytosolic amounts and (2) to remove laforin from glycogen as soon as laforin has removed its target phosphate. In the absence of malin, excessive amounts of laforin bind glycogen and, like excess phosphate, render glycogen precipitation- and aggregation-prone, digestion-resistant, and poorly branched, leading to the formation of LBs. Additionally, accumulation of laforin in glycogen and then in LBs depletes the cytosol of free laforin, leading to a secondary laforin deficiency [64, 65•, 66, 67•].
Epm2a −/− null mice are developmentally normal but show behavioral abnormalities at 4 months of age and signs of ataxia, and myoclonus epilepsy at 9 months. At 2 months, homozygous null mutants develop widespread degeneration of neurons. Swelling of mitochondria, endoplasmic reticulum, and Golgi apparatus with lysis of cell membranes leads to cell death. As LBs become more prominent at 4–12 months, organelles and nuclei are disrupted [66].
The presence of neurodegeneration prior to the formation of LBs is explained by the role of laforin in autophagosome formation and thus in the clearance of misfolded proteins in neuronal cells [68, 69].
AMP-Activated Protein Kinase
The AMPK complex, which acts as a sensor of cellular energy status, is a heterotrimer composed of a catalytic α subunit and regulatory β and γ subunits [70]. The γ subunits act in pairs to form two binding sites for the regulatory nucleotides AMP and ATP [71]. Once activated by cellular stresses that deplete ATP and raise the concentration of AMP, AMPK switches on various ATP-producing catabolic pathways and, conversely, downregulates ATP-consuming processes [72].
Mutations in the PRKAG2 gene, encoding the γ2 subunit isoform of AMPK, give rise to a moderate, essentially heart-specific, glycogenosis with clinical onset typically in late adolescence or in the third decade of life, ventricular preexcitation predisposing to supraventricular arrhythmias, mild-to-severe cardiac hypertrophy, enhanced risk of sudden cardiac death in midlife, and autosomal dominant inheritance with full penetrance (familial hypertrophic cardiomyopathy with Wolff–Parkinson–White syndrome) [27].
Notably, Burwinkel et al. [27] identified a single dominant mutation (R531Q) in PRKAG2 in three patients with a fatal infantile heart glycogenosis, initially and erroneously attributed to PHK deficiency . Later, Akman et al. [73] found a second mutation (R384T) in another infant with the same disorder .
From a pathogenetic perspective, AMPK is known to lead to an increase in the phosphorylation and inactivation of GS. Moreover, it positively regulates the formation of the laforin–malin complex, thus inducing PTG proteasomal degradation.
Transgenic mice overexpressing the PRKAG2 N488I missense mutation under control of the cardiac-specific α-myosin heavy chain show cardiomyopathy, cardiac glycogen accumulation in the annulus fibrosus resulting in remodeling of atrioventricular conduction pathways, and electrophysiological abnormalities similar to the ones described in patients harboring the same mutation [74].
Finally, Costford et al. [75] identified the rare mutation R225W in the gene PRKAG3 encoding the AMPK regulatory γ3 subunit. This isoform is expressed exclusively in skeletal muscle, and its defect results in 90 % increase in glycogen level, and an approximate 30 % decrease in the level of intramuscular triglyceride.
Conclusions
Our knowledge of the genetics, molecular biology, and biochemistry of glycogen disorders is continuously expanding. Indeed, new coordinated signaling pathways which regulate the actions of the enzymes directly involved in glycogen metabolism have been discovered and are being progressively clarified (laforin–malin, AMPK). Moreover, lysosomal disposal of glycogen is now being revisited to elucidate the molecular mechanisms of transport of glycogen to lysosomes. The generation of murine models, which successfully recapitulate the clinical, histological, and biochemical aspects of the different glycogenoses has played a crucial role. These experimental models allow analysis of early histopathological changes that can define the primary events of the diseases (e.g., congenital cardiomyopathies or Lafora disease). Moreover, they can be used to test new genetic and pharmacological therapeutic strategies.
References
Papers of particular interest, published recently have been heighted as: • Of important •• Of major important
Roach PJ. Glycogen and its metabolism. Curr Mol Med. 2002;2:101–20.
DiMauro S, Spiegel R. Progress and problems in muscle glycogenoses. Acta Myol. 2011;30:96–102.
DiMauro S, Garone C. Metabolic disorders of fetal life: glycogenoses and mitochondrial defects of the mitochondrial respiratory chain. Semin Fetal Neonatal Med. 2011;16:181–9.
Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem J. 2012;441:763–87.
van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372:1342–53.
Moslemi AR, Lindberg C, Nilsson J, et al. Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. N Engl J Med. 2010;362:1203–10.
Nilsson J, Halim A, Moslemi AR, et al. Molecular pathogenesis of a new glycogenosis caused by a glycogenin-1 mutation. Biochim Biophys Acta. 2012;1822:493–9.
Kollberg G, Tulinius M, Gilljam T, et al. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N Engl J Med. 2007;357:1507–14.
Pederson BA, Chen H, Schroeder JM, et al. Abnormal cardiac development in the absence of heart glycogen. Mol Cell Biol. 2004;24:7179–87.
Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med. 2002;2:177–88.
Bruno C, Cassandrini D, Assereto S, et al. Neuromuscular forms of glycogen branching enzyme deficiency. Acta Myol. 2007;26:75–8.
Magoulas PL, El-Hattab AW, Roy A, et al. Diffuse reticuloendothelial system involvement in type IV glycogen storage disease with a novel GBE1 mutation: a case report and review. Hum Pathol. 2012;43:943–51.
Bruno C, van Diggelen OP, Cassandrini D, et al. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology. 2004;63:1053–8.
Reusche E, Aksu F, Goebel HH, et al. A mild juvenile variant of type IV glycogenosis. Brain Dev. 1992;14:36–43.
Servidei S, Riepe RE, Langston C, et al. Severe cardiopathy in branching enzyme deficiency. J Pediatr. 1987;111:51–516.
Goebel HH, Shin YS, Gullotta F, et al. Adult polyglucosan body myopathy. J Neuropathol Exp Neurol. 1992;51:24–35.
Lossos A, Barash V, Soffer D, et al. Hereditary branching enzyme dysfunction in adult polyglucosan body disease: a possible metabolic cause in two patients. Ann Neurol. 1991;30:655–62.
Bruno C, Servidei S, Shanske S, et al. Glycogen branching enzyme deficiency in adult polyglucosan body disease. Ann Neurol. 1993;33:88–93.
Taratuto AL, Akman HO, Saccoliti M, Riudavets M, Arakaki N, Mesa L, Sevlever G, Goebel H, DiMauro S. Branching enzyme deficiency/glycogenosis storage disease type IV presenting as a severe congenital hypotonia: muscle biopsy and autopsy findings, biochemical and molecular genetic studies. Neuromuscul Disord. 2010;20:783–90.
Fyfe JC, Giger U, Van Winkle TJ, et al. Glycogen storage disease type IV: inherited deficiency of branching enzyme activity in cats. Pediatr Res. 1992;32:719–25.
Ward TL, Valberg SJ, Adelson DL, et al. Glycogen branching enzyme (GBE1) mutation causing equine glycogen storage disease IV. Mamm Genome. 2004;15:570–7.
Lee YC, Chang CJ, Bali D, et al. Glycogen-branching enzyme deficiency leads to abnormal cardiac development: novel insights into glycogen storage disease IV. Hum Mol Genet. 2011;20:455–65.
Akman HO, Sheiko T, Tay SK, et al. Generation of a novel mouse model that recapitulates early and adult onset glycogenosis type IV. Hum Mol Genet. 2011;20:4430–9.
• Goldstein J, Austin S, Kishnani P, Bali D. Phosphorylase kinase deficiency. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews™ [Internet]. Seattle: University of Washington; 1993–2011 May 31. This is a comprehensive review of physiopathological and clinical features of glycogenosis type VIII.
Wehner M, Clemens PR, Engel AG, Kilimann MW. Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit. Hum Mol Genet. 1994;3:1983–7.
Bruno C, Manfredi G, Andreu AL, et al. A splice junction mutation in the alpha(M) gene of phosphorylase kinase in a patient with myopathy. Biochem Biophys Res Commun. 1998;249:648–51.
Burwinkel B, Hu B, Schroers A, et al. Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. Eur J Hum Genet. 2003;11:516–26.
Wuyts W, Reyniers E, Ceuterick C, Storm K, de Barsy T, Martin JJ. Myopathy and phosphorylase kinase deficiency caused by a mutation in the PHKA1 gene. Am J Med Genet A. 2005;133:82–4.
Echaniz-Laguna A, Akman HO, et al. Muscle phosphorylase b kinase deficiency revisited. Neuromuscul Disord. 2010;20:125–7.
Ørngreen MC, Schelhaas HJ, Jeppesen TD, et al. Is muscle glycogenolysis impaired in X-linked phosphorylase b kinase deficiency? Neurology. 2008;70:1876–82.
Preisler N, Orngreen MC, Echaniz-Laguna A, et al. Muscle phosphorylase kinase deficiency: a neutral metabolic variant or a disease? Neurology. 2012;78:265–8.
Arenas J, Martín MA, Andreu AL. Glycogen storage disease type V. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews™ [Internet]. Seattle: University of Washington; 1993–2006 Apr 19 (updated 12 May 2009).
Lucia A, Ruiz JR, Santalla A, et al. Genotypic and phenotypic features of McArdle disease: insights from the Spanish national registry. J Neurol Neurosurg Psychiatry. 2012;83:322–8.
Nogales-Gadea G, Rubio JC, Fernandez-Cadenas I, et al. Expression of the muscle glycogen phosphorylase gene in patients with McArdle disease: the role of nonsense-mediated mRNA decay. Hum Mutat. 2008;29:277–83.
Bruno C, Cassandrini D, Martinuzzi A, et al. McArdle disease: the mutation spectrum of PYGM in a large Italian cohort. Hum Mutat. 2006;27:718.
Vissing J, Duno M, Schwartz M, Haller RG. Splice mutations preserve myophosphorylase activity that ameliorates the phenotype in McArdle disease. Brain. 2009;132:1545–52.
Quinlivan R, Buckley J, James M, et al. McArdle disease: a clinical review. J Neurol Neurosurg Psychiatry. 2010;81:1182–8.
Haller RG, Vissing J. Spontaneous “second wind” and glucose-induced second “second wind” in McArdle disease: oxidative mechanisms. Arch Neurol. 2002;59:1395–402.
• Lucia A, Nogales-Gadea G, Perez M, et al. McArdle disease: what do neurologists need to know? Nat Clin Pract Neurol. 2008;4:568–77. This is a comprehensive review of clinical features and management aspects of glycogenosis type V.
Angelos S, Valberg SJ, Smith BP, et al. Myophosphorylase deficiency associated with rhabdomyolysis and exercise intolerance in 6 related charolais cattle. Muscle Nerve. 1995;18:736–40.
Tan P, Allen JG, Wilton SD, et al. A splice-site mutation causing ovine McArdle's disease. Neuromuscul Disord. 1997;7:336–42.
Nogales-Gadea G, Pinós T, Lucia A, et al. Knock-in mice for the R50X mutation in the PYGM gene present with McArdle disease. Brain. 2012;135:2048–57.
• Kishnani PS, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12:446–63. This is a comprehensive review of physiopathological and clinical features of glycogenosis type III.
Goldstein JL, Austin SL, Boyette K, et al. Molecular analysis of the AGL gene: identification of 25 novel mutations and evidence of genetic heterogeneity in patients with glycogen storage disease type III. Genet Med. 2010;12:424–30.
Vertilus SM, Austin SL, Foster KS, et al. Echocardiographic manifestations of glycogen storage disease III: increase in wall thickness and left ventricular mass over time. Genet Med. 2010;12:413–23.
Gregory BL, Shelton GD, Bali DS, Chen YT, Fyfe JC. Glycogen storage disease type IIIa in curly-coated retrievers. J Vet Intern Med. 2007;21:40–6.
Yi H, Thurberg BL, Curtis S, et al. Characterization of a canine model of glycogen storage disease type IIIa. Dis Model Mech. 2012;5:804–11.
Nakajima H, Raben N, Hamaguchi T, Yamasaki T. Phosphofructokinase deficiency; past, present and future. Curr Mol Med. 2002;2:197–212.
Toscano A, Musumeci O. Tarui disease and distal glycogenoses: clinical and genetic update. Acta Myol. 2007;26:105–7.
Haller RG, Vissing J. No spontaneous second wind in muscle phosphofructokinase deficiency. Neurology. 2004;62(1):82–6.
Musumeci O, Bruno C, Mongini T, et al. Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). Neuromuscul Disord. 2012;22:325–30.
García M, Pujol A, Ruzo A, et al. Phosphofructo-1-kinase deficiency leads to a severe cardiac and hematological disorder in addition to skeletal muscle glycogenosis. PLoS Genet. 2009;5:e1000615.
Beutler E. PGK deficiency. Br J Haematol. 2007;136:3–11.
Spiegel R, Area Gomez E, Akman HO, et al. Myopathic form of phosphoglycerate kinase (PGK) deficiency: a new case and pathogenic considerations. Neuromusc Disord. 2009;19:207–11.
Sotiriou E, Greene P, Krishna S, et al. Myopathy and parkinsonism in phosphoglycerate kinase deficiency. Muscle Nerve. 2010;41:707–10.
Naini A, Toscano A, Musumeci O, et al. Muscle phosphoglycerate mutase deficiency revisited. Arch Neurol. 2009;66:394–8.
Kanno T, Maekawa M. Lactate dehydrogenase M-subunit deficiencies: clinical features, metabolic background, and genetic heterogeneities. Muscle Nerve. 1995;3:S54–60.
Kreuder J, Borkhardt A, Repp R, et al. Inherited metabolic myopathy and hemolysis due to a mutation in aldolase a. N Engl J Med. 1996;334:1100–4.
Comi GP, Fortunato F, Lucchiari S, et al. Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Ann Neurol. 2001;50:202–7.
•• Stojkovic T, Vissing J, Petit F, et al. Muscle glycogenosis due to phosphoglucomutase 1 deficiency. N Engl J Med. 2009;361:425–7. The is the first description of this new glycogen disorder.
Delgado-Escueta AV. Advances in Lafora progressive myoclonus epilepsy. Curr Neurol Neurosci Rep. 2007;7:428–33.
Minassian BA, Lee JR, Herbrick JA, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20:171–4.
Vilchez D, Ros S, Cifuentes D, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10:1407–13.
DePaoli-Roach AA, Tagliabracci VS, Segvich DM, et al. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to Lafora bodies and the accumulation of insoluble laforin. J Biol Chem. 2010;285:25372–81.
• Turnbull J, Wang P, Girard JM, et al. Glycogen hyperphosphorylation underlies Lafora body formation. Ann Neurol. 2010;68:925–33. This article defines the relevance of glycogen phosphate accumulation in a mouse model of malin deficiency.
Ganesh S, Delgado-Escueta AV, Sakamoto T, et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. 2002;11:1251–62.
• Tagliabracci VS, Turnbull J, Wang W, et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci USA. 2007;104:19262–6. This article defines the relevance of glycogen phosphate accumulation in a mouse model of laforin deficiency.
Puri R, Ganesh S. Autophagy defects in Lafora disease: cause or consequence? Autophagy. 2012;8:289–90.
Delgado-Escueta AV. Lafora progressive myoclonus epilepsy: glycogen storage disease vs neurodegenerative disease. Neurology. 2012;79:21–2.
Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–908.
Scott JW, Norman DG, Hawley SA, Kontogiannis L, Hardie DG. Protein kinase substrate recognition studied using the recombinant catalytic domain of AMP-activated protein kinase and a model substrate. J Mol Biol. 2002;317:309–23.
Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002;109:357–62.
Akman HO, Sampayo JN, Ross FA, et al. Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase. Pediatr Res. 2007;62:499–504.
Arad M, Moskowitz IP, Patel VV, et al. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-white syndrome in glycogen storage cardiomyopathy. Circulation. 2003;107:2850–6.
Costford SR, Kavaslar N, Ahituv N, et al. Gain-of-function R225W mutation in human AMPKgamma(3) causing increased glycogen and decreased triglyceride in skeletal muscle. PLoS One. 2007;2:e903.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Nerve and Muscle
Rights and permissions
About this article
Cite this article
Gazzerro, E., Andreu, A.L. & Bruno, C. Neuromuscular Disorders of Glycogen Metabolism. Curr Neurol Neurosci Rep 13, 333 (2013). https://doi.org/10.1007/s11910-012-0333-0
Published:
DOI: https://doi.org/10.1007/s11910-012-0333-0
Keywords
- Glycogen metabolism
- Glycogen storage disease
- Glycogenoses
- Cardiomyopathy
- Vacuolar myopathy
- Exercise intolerance
- Weakness
- Glycogenin
- Glycogen synthase
- Branching enzyme
- Debrancher
- Myophosphorylase
- Phosphorylase b kinase
- Phosphofructokinase
- Phosphoglycerate kinase
- Phosphoglycerate mutase
- Danon disease
- Laforin
- Malin
- Polyglucosan
- Lafora bodies
- Autophagy
- Second wind phenomenon
- AMP-activated protein kinase