
Abstract
New epilepsy treatments are needed that not only inhibit seizures symptomatically (antiseizure) but also prevent the development of epilepsy (antiepileptogenic). The mammalian target of rapamycin (mTOR) pathway may mediate mechanisms of epileptogenesis and serve as a rational therapeutic target. mTOR inhibitors have antiepileptogenic and antiseizure effects in animal models of the genetic disease, tuberous sclerosis complex. The mTOR pathway is also implicated in epileptogenesis in animal models of acquired epilepsy and infantile spasms, although the effects of mTOR inhibitors are variable depending on the specific conditions and model. Furthermore, beneficial effects on seizures are lost when treatment is withdrawn, suggesting that mTOR inhibitors are “epileptostatic” in only stalling epilepsy progression during treatment. Clinical studies of rapamycin in human epilepsy are limited, but suggest that mTOR inhibitors at least have antiseizure effects in tuberous sclerosis patients. Further studies are needed to assess the full potential of mTOR inhibitors for epilepsy treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epilepsy is a common, neurological disorder affecting approximately 1 % of the population. To date, the treatment of epilepsy has generally been unsatisfactory with approximately one third of individuals refractory to medication. In addition, with the possible exception of epilepsy surgery, current treatments ameliorate seizures symptomatically (ie, antiseizure), with no effect on the underlying pathophysiology or course of the disease itself. Therefore, the ability to develop new epilepsy treatments that alter or prevent the development of epilepsy (ie, antiepileptogenic) represents a holy grail in epilepsy research.
Epileptogenesis is the process leading to the development of epilepsy following a brain injury or defect from environmental or genetic causes. After the initial insult, epileptogenesis results from a complex interplay of multiple factors during a latent period typically lasting months to years, ultimately culminating in neuronal hyperexcitability and the expression of seizures. Multiple model systems have been developed to study epileptogenesis. Perhaps the most relevant models are those that mimic common causes of acquired epilepsy in humans, such as following traumatic brain injury (TBI) or status epilepticus. Among genetic causes of epilepsy, tuberous sclerosis complex (TSC) is an attractive model given its high propensity for seizures and the possibility of early detection via genetic testing and non-neurological findings. TSC, an autosomal-dominant disorder due to mutations in the TSC1 (hamartin) or TSC2 (tuberin) genes, affects multiple organ systems, but neurological involvement, especially epilepsy, usually accounts for the most disabling symptoms [1]. The exciting discovery that the hamartin and tuberin proteins limit activation of the mammalian target of rapamycin (mTOR) signaling pathway sparked a series of discoveries that are relevant not only to epileptogenesis in TSC, but potentially also to more common, acquired epilepsies.
This review aims to discuss the role that the mTOR pathway plays in epileptogenesis and how inhibition of this pathway has potential for epilepsy treatment. While other works have also covered this subject [2–4], this review analyzes the most recent up-to-date animal and human studies, including some published in preliminary form, related to mTOR inhibitors and epilepsy.
The mTOR Pathway
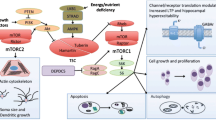
mTOR is a critical protein kinase that functions to integrate multiple intra- and extracellular signals to regulate cell growth, metabolism, proliferation, and survival via alterations in gene expression and protein translation (Fig. 1). The complex details of mTOR biology have recently been reviewed elsewhere and are outside the scope of this review [4, 5]. In brief, mTOR forms two complexes, mTORC1, which can be inhibited by rapamycin, and mTORC2, which is largely rapamycin-insensitive. mTORC1 activates a number of downstream pathways including stimulation of mRNA translation via activation of the p70 ribosomal S6 kinase 1 (S6K1) and the eukaryotic initiation factor 4E binding protein-1 (4E-BP1). These actions mediate many of the functional effects of the mTOR pathway via modulation of protein synthesis.

Regulation of the mTOR signaling pathway. The serine-threonine protein kinase, mTOR, forms two complexes, mTORC1, which is rapamycin-sensitive, and mTORC2 (not shown). The mTOR pathway activates downstream signaling mechanisms primarily involved in regulating protein synthesis related to multiple functions, such as cell growth and proliferation, as well as other processes that may relate directly to epileptogenesis. In turn, the mTOR pathway may be activated or inhibited by various physiological or pathological stimuli via various upstream signaling pathways and intermediary proteins (TSC1, TSC2, Rheb). AMPK—5′ adenosine monophosphate-activated protein kinase; eIF4E—elongation initiation factor 4E; ERK—extracellular signal-regulated kinases; IGF—insulin-like growth factor; mTOR—mammalian target of rapamycin; PI3K—phosphatidylinositol-3 kinase; PTEN—phosphatase and tensin homolog deleted on chromosome 10; Rheb—Ras homolog enriched in brain; STRADα—STE20-related kinase adapter-α; S6—ribosomal protein S6; S6K—ribosomal S6 kinase; TSC1—tuberous sclerosis complex 1 protein; TSC2—tuberous sclerosis complex 2 protein; 4E-BP1—elongation factor 4E binding protein-1
Hamartin and tuberin act as a complex upstream of mTOR and inhibit the mTOR pathway via inhibition of Rheb (Ras homolog enriched in brain) (Fig. 1). Multiple upstream pathways stimulate or inhibit the mTOR pathway by interacting with the hamartin and tuberin complex to control important physiological functions. For example, regulation of energy metabolism in response to conditions promoting growth or starvation is achieved through opposite effects on the hamartin-tuberin complex via growth factor stimulation of the phosphatidylinositol-3 kinase (PI3K) pathway or energy deprivation leading to LKβ1/AMPK pathway activation. Several interesting findings further support the role of this pathway in regulation of energy and growth that may relate to epilepsy. The high-fat, low-carbohydrate, ketogenic diet is a well-established treatment for epilepsy [6], but the mechanism of action is unknown. Interestingly, rats fed a ketogenic diet demonstrated reduction of both upstream and downstream mTOR pathway markers, suggesting that the mTOR pathway may be involved in effects of the ketogenic diet on growth and seizures [7]. The link between mTOR and the role of nutrient signaling is further elucidated in patients with a rare disorder known as polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE), in which deletions in the STRADA gene cause dysregulation of mTOR signaling via a reduction in the LKβ1/AMPK pathway [8].
Probably the strongest link between mTOR and epilepsy occurs in TSC, one of the most common genetic causes of epilepsy. In TSC, the result of inactivating mutations in TSC1 or TSC2 is to reduce inhibition of the mTOR pathway and therefore hyperactivate downstream targets of mTOR [1]. Abnormal mTOR signaling was initially implicated in causing abnormal cell growth and proliferation contributing to cortical malformations (eg, tubers) and brain tumors (eg, astrocytomas) in TSC [9]. This finding has led to the establishment of mTOR inhibitors for treating astrocytoma growth in TSC patients [10•]. Intriguingly, some forms of focal cortical dysplasias, specifically type IIB, which resemble cortical tubers histologically and represent a common cause of intractable epilepsy, also exhibit abnormalities in TSC gene polymorphisms, hamartin and tuber expression, and other mTOR markers [11–19]. Thus, a strong association between the mTOR pathway and epilepsy has been found in several types of human epilepsy. However, evidence supporting a causal role of mTOR in epileptogenesis is less clear and primarily derives from animal models of epilepsy.
mTOR and Epileptogenesis in Animal Models
Genetic Epilepsies
TSC
Multiple mouse models of TSC have been generated by specific disruption of the Tsc1 [20, 21, 22•, 23, 24] or Tsc2 genes [25–27] in neurons, glia, or brain progenitor cells (Table 1). These models recapitulate many aspects of the central nervous system phenotype found in TSC, including epilepsy and deficits in learning and memory. At the cellular level, these animal models also exhibit abnormalities found in human TSC brain specimens, such as enlarged and dysplastic neurons or vacuolated giant cells, abnormal neuronal organization, reduced myelin, and astrogliosis. Although the precise molecular mechanisms causing epileptogenesis in TSC are not known, specific molecular defects, such as in astrocyte glutamate transporters, have been identified and may contribute to epileptogenesis in these mouse models.
A critical role of the mTOR pathway in epileptogenesis has been firmly established in many of these TSC models (Table 1). The brain abnormalities are associated with hyperactivation of the mTOR pathway and postnatal treatment with mTOR inhibitors decreased megalencephaly, neuronal hypertrophy, and astrogliosis. Postnatal treatment with mTOR inhibitors decreased seizures when started after the onset of seizures (ie, antiseizure) [28] or prevented the development of epilepsy when treatment was initiated prior to the onset of seizures (ie, antiepileptogenic) [22•, 23, 27–29]. Rapamycin also improved the cognitive deficits seen in Tsc2 mutant mice [26]. Unfortunately, neither the antiseizure nor antiepileptogenic benefits persisted in any of the TSC mutant mouse models once rapamycin was withdrawn, indicating that chronic, indefinite treatment was necessary to maintain efficacy.
Pten Models
An additional window into the role that abnormal activation of the mTOR pathway plays in genetic epilepsies arises from study of Pten (phosphatase and tensin homolog deleted on chromosome ten) mutant animal models. Pten is a tumor suppressor gene that exerts its effects on growth via modulation of the PI3K/AKT pathway, which is an upstream regulator of TSC1 and TSC2 (Fig. 1). Various human diseases with Pten mutations have been identified involving tumor or hamartoma growth in various organs. In addition, a common theme of some of these diseases is the presence of seizures [30].
A couple of Pten knockout mice have been developed targeting the central nervous system. In one mouse model, Pten inactivation in postnatal granule neurons of the dentate gyrus and cerebellum led to megalencephaly, neuronal hypertrophy, seizures, and premature death [31, 32]. Treatment with mTOR inhibitors decreased seizures and associated pathological abnormalities and prolonged life [33, 34]. Similar to the TSC models, seizures returned within weeks of cessation of rapamycin therapy, although subsequent intermittent rapamycin treatment was able to maintain a long-term antiseizure effect [35•]. In another, similar Pten mouse model, rapamycin not only decreased seizures and neuronal hypertrophy, but also reversed deficits in social behavior suggestive of autism [36].
Acquired Epilepsies
While the link between certain genetic diseases, especially TSC, and the mTOR pathway is well-established, these genetic diseases are relatively rare causes of epilepsy. Environmental insults or other acquired brain disorders represent more common etiologies of epilepsy. Thus, a question of potentially broader clinical relevance and impact is whether abnormal mTOR signaling may contribute to various epilepsies due to acquired brain injury. In a general scheme of epileptogenesis in acquired epilepsy, the initial precipitating injury triggers epileptogenic mechanisms during a latent period, which eventually leads to the development of spontaneous seizures. In theory, activation of the mTOR pathway could represent one of the initial signals during the latent period, which trigger a number of downstream cellular and molecular changes in the brain that ultimately cause increased neuronal excitability and seizure generation. There is now evidence that the mTOR pathway is indeed activated following various types of brain injuries in animal models. Furthermore, in some models of acquired epilepsy, mTOR appears to mediate cellular abnormalities implicated in epileptogenesis, such as mossy fiber sprouting in the hippocampus, as well as possibly contributing to seizure generation itself. Experimental evidence for the involvement of the mTOR pathway in animal models of different types of acquired epilepsy are briefly reviewed in sequence, and are also summarized in Table 2.
Status Epilepticus Injury Models
Some of the most popular animal models of epilepsy involve an initial episode of status epilepticus, triggered by drugs or electrical stimulation, which then causes brain injury and subsequent development of spontaneous seizures. The mechanisms of epileptogenesis in these status epilepticus models are still debated, but may involve neuronal death, synaptic reorganization (eg, mossy fiber sprouting), neurogenesis, and molecular changes in ion channels. The mTOR pathway has been clearly shown to be activated during and following status epilepticus in both the kainate and pilocarpine models [37•, 38, 39]. Furthermore, mTOR almost certainly mediates some of the cellular changes that have been implicated in epileptogenesis, particularly mossy fiber sprouting, as mTOR inhibitors consistently reduce the degree of mossy fiber sprouting using different models and paradigms [37•, 38, 39]. Interestingly, however, the effects of mTOR inhibition on mossy fiber sprouting require continued treatment, as withdrawal of rapamycin leads to reemergence of mossy fiber sprouting [38]. The mTOR pathway may also be involved in neuronal death and neurogenesis following status epilepticus, although this appears to be dependent on the specific timing and model [37•].
The effect of mTOR inhibitors on epilepsy itself in the status epilepticus models is more complicated and variable. In the kainate model in rats, rapamycin treatment shortly before or after status epilepticus decreases the subsequent development of epilepsy [37•], suggesting a possible antiepileptogenic effect. However, in the pilocarpine model, late rapamycin treatment in rats that have already developed spontaneous epilepsy can significantly reduce seizure frequency, more consistent with a direct antiseizure effect [39]. In contrast, in another study involving the pilocarpine model in mice, early rapamycin did not appear to have an antiepileptogenic effect for preventing epilepsy; however, the concurrent late use of rapamycin in the control group during the monitoring period to control for anticonvulsant effects does not preclude the possibility of an undetected short-term antiseizure effect [40•]. Furthermore, another electrical stimulation model of the amygdala did not find an effect of rapamycin on subsequent development of epilepsy, but since rapamycin was stopped before the end of the monitoring period, the possibility of an undetected transient short-term effect also exists [41]. Other explanations for variation in these studies may involve differences in species, dose, and timing of the effects, as well as electroencephalography (EEG) monitoring methods. At a minimum, the status epilepticus models demonstrate the complexity of mTOR’s involvement in epileptogenesis and ictogenesis and raise the question as to whether the effects of mTOR inhibitors are more consistent with antiepileptogenic or antiseizure mechanisms [42].
Traumatic Brain Injury Models
TBI and posttraumatic epilepsy represent one of the most common and important types of human epilepsy. Furthermore, antiepileptogenic drug trials, attempting to prevent posttraumatic epilepsy, have been successfully designed and conducted, although the results have been negative [43]. Similar to status epilepticus models, abnormal activation of the mTOR pathway has been demonstrated in several animal models of TBI [44–47]. Rapamycin decreases neuronal death and functional deficits, including spatial memory impairment, following TBI [45, 47]. While effects of mTOR inhibitors on posttraumatic epilepsy have not yet been rigorously tested, preliminary data suggest that rapamycin does decrease the development of posttraumatic epilepsy and associated neuronal death and mossy fiber sprouting in the controlled cortical impact model in mice [46], as well as in an in vitro organotypic culture model of posttraumatic epileptogenesis [48]. If these preliminary results are confirmed, this would have direct clinical implications for patients with TBI.
Infantile Spasms Models
Infantile spasms represent a characteristic epilepsy syndrome of infancy and early childhood, characterized by epileptic spasms, developmental delay, and hypsarrhythmia on EEG and are usually associated with a poor prognosis. Infantile spasms can be caused by a variety of different brain insults during early development or can be of unknown cause (“cryptogenic”). Recently a number of animal models of infantile spasms have been developed and characterized and the potential involvement of the mTOR pathway has been tested in a couple of them. In a “multiple-hit” model involving several pharmacologically induced “injuries” to model symptomatic infantile spasms, rapamycin can decrease the frequency of pre-existing spasms in neonatal rats [49•], suggesting a direct antiseizure effect. In addition, rapamycin treatment decreased the subsequent development of cognitive deficits observed in this model, indicating a disease-modifying effect. Interestingly, however, rapamycin did not decrease the development of other non-spasm seizure types, indicating that mTOR may only be involved in mediating specific types of seizures in this animal model. Furthermore, in another model of “cryptogenic” spasms, rapamycin appeared to have no direct effect on the spasms [50]. Again differences in effects of rapamycin in different models suggest that the role of the mTOR pathway in epileptogenesis may vary depending on the type and cause of epilepsy and the timing of rapamycin administration.
Other Models
Preliminary studies have also investigated the potential role of mTOR in other types of acquired epilepsy models. In a model of neonatal hypoxic brain injury, acute hypoxia induces immediate seizures in neonatal rats, which later develop a decreased seizure threshold and spontaneous seizures in adulthood. Preliminary studies indicate that the mTOR pathway is hyperactivated following neonatal hypoxia and rapamycin inhibits the subsequent change in seizure threshold, suggesting an antiepileptogenic effect [51]. In a model of rapid electrical kindling, repetitive electrical stimulation leads to increasing epileptiform discharges. In a limited number of experiments, rapamycin was reported to decrease the development of epileptiform activity in this model [52].
Data from Human Studies
Given the reports from animal models, there is burgeoning excitement to further assess the role that inhibition of the mTOR pathway might play in the treatment of clinical epilepsy in humans. Although this would ideally be assessed for its antiepileptogenic potential for preventing epilepsy in high-risk patients, there are clear barriers to designing and conducting antiepileptogenesis trials in people. The limited data available to date have only assessed the role of mTOR inhibition as an antiseizure medication in patients with established, intractable epilepsy.
The human data that exist come almost exclusively from TSC patients. A single case report initially suggested a beneficial effect of rapamycin in reducing seizure frequency in a child with TSC and intractable epilepsy [53]. The largest available human data derive from a phase II prospective, open-label trial of everolimus (a rapamycin analog) that was designed primarily to assess the efficacy of everolimus in inhibiting growth of subependymal giant cell astrocytomas in patients with TSC [10•]. Seizure frequency was a secondary end point of the study, as assessed by self-reports of seizure frequency, as well as 24-hour video EEG at baseline and after 6 months of treatment. Twenty-eight patients with TSC were enrolled ranging in age from 3 to 34 years and received oral everolimus (3 mg/m2/d, serum levels 5–15 ng/mL). Treatment with everolimus resulted in a significant decrease in self-reported seizure frequency. Sixteen patients underwent video EEG evaluation with 9 of 10 patients with active seizures demonstrating a decrease in seizure frequency and the 10th patient demonstrating an increase in seizure frequency. The remaining six patients had no change in seizure frequency but five of these were seizure free both at baseline and in follow-up.
These intriguing results have sparked a subsequent clinical trial of everolimus with seizures as the primary end point. Preliminary data from this study were recently presented in abstract form [54]. In this open-label, multicenter combined phase I/II clinical trial, patients greater than 2 years old with medically refractory epilepsy secondary to TSC were treated with everolimus (5 mg/m2/day, 5–15 ng/mL). Seizures were assessed by seizure diary and 24-hour video EEG during the baseline and at the end of an 8-week treatment period. Fifteen of 17 total patients reported at least a 25 % reduction in seizure frequency with nine patients reporting greater than 50 % reduction. One patient reported an increase in seizure frequency.
Conclusions
There is now a myriad of data, mostly from animal models, but also from limited human studies, supporting a role of the mTOR pathway in epilepsy and suggesting that mTOR inhibitors may represent rational therapeutic agents. However, the specific contexts and mechanisms involving the mTOR pathway and the type of therapeutic action of mTOR inhibitors are uncertain and may vary depending on a number of factors, such as timing of treatment, seizure type, and epilepsy syndrome. The main issue is whether the mTOR pathway is involved in epileptogenesis (ie, the development of epilepsy) or the maintenance of epilepsy/ictogenesis (ie, seizure generation) and correspondingly whether mTOR inhibitors primarily have antiepileptogenic (ie, preventing the development or progression of epilepsy) or antiseizure actions (ie, suppressing the symptoms of seizures in patients who already have epilepsy).
At this point, there is evidence that mTOR inhibitors may have both antiepileptogenic and antiseizure actions, again depending on the context and type/model of epilepsy. In theory, antiepileptogenic and antiseizure effects may have completely separate and unique mechanisms of action. However, a distinct separation between antiepileptogenic and antiseizure mechanisms may be artificial. Given the diversity of cellular and molecular processes that may be regulated by the mTOR pathway, it is possible that mTOR inhibitors could have a range of overlapping actions spanning both antiepileptogenic and antiseizure effects. Plausible antiepileptogenic actions of mTOR inhibition include regulation of synaptic plasticity, neuronal death, neurogenesis, and long-term actions on metabolism and protein synthesis. In addition, while there is minimal evidence that mTOR inhibitors have immediate, direct effects on neuronal excitability akin to traditional anticonvulsant drugs [55, 56], mTOR inhibition could regulate protein synthesis of ion channels, neurotransmitter receptors, or other proteins involved in neuronal signaling, resulting in antiseizure effects in patients with established epilepsy.
Regardless of mechanism, one key principle that has emerged from the animal studies is the importance of the timing and duration of treatment with mTOR inhibitors. In the TSC models, mTOR inhibitors consistently have apparent antiepileptogenic actions in preventing epilepsy when treatment is initiated early; however, when treatment is then discontinued, epilepsy subsequently emerges [22•, 28, 29]. Thus, mTOR inhibitors are not a cure for the epilepsy found in TSC and persistent, lifelong treatment may be required to maintain effectiveness. The requirement for long-term treatment is not surprising for a genetic disease like TSC, as the underlying genetic defect persists and becomes unmasked upon rapamycin discontinuation, allowing reactivation of the mTOR pathway. In theory, acquired epilepsies may not have a requirement for long-term treatment, as some epileptogenic mechanisms may only occur for a limited, critical period after the initial brain injury. Interestingly, however, there is some evidence that discontinuation of rapamycin treatment also leads to loss of effectiveness, at least for suppression of late seizures and mossy fiber sprouting in the pilocarpine model of acquired epilepsy [38, 39]. Although not specifically examined, a requirement for persistent mTOR inhibition might also explain variability in effects on seizures among various models of acquired seizures, including status epilepticus and infantile spasms models [37•,39, 40•, 41, 49, 50]. In summarizing rapamycin’s actions, Galanopoulou et al. [57] proposed that mTOR inhibitors may have “epileptostatic” properties that are intermediate between antiepileptogenic and antiseizure actions, with the potential to prevent epilepsy but the requirement for chronic treatment to maintain efficacy.
The clinical data in people are much more limited, only involving ongoing antiseizure trials in TSC patients with intractable epilepsy. If the current clinical trials definitively demonstrate an antiseizure effect of mTOR inhibitors, a logical next step would involve a true antiepileptogenic trial. Antiepileptogenic drug trials have been attempted in the past, primarily involving conventional antiseizure medications in patients with TBI, but the results have been negative [43]. Patients with TSC may be an appropriate initial target population for an antiepileptogenic trial with mTOR inhibitors for several reasons including the high rate of epilepsy, the medical refractory nature of epilepsy in TSC patients, and the ability to diagnosis TSC prior to epilepsy onset and in many cases prenatally. However, there are many potential barriers to an antiepileptogenic trial. Although the rate of epilepsy is quite high, it is not 100 % even in this population and to date there are no clearly identifiable predictors of epilepsy in TSC. As such, treatment of all patients with TSC would needlessly expose some patients to side effects without clear benefit. Additionally, the timing of treatment initiation is not clear. One could argue that prenatal treatment of women carrying fetuses with TSC identified by genetic testing or prenatal ultrasound may be necessary to attain the full benefit of rapamycin treatment since cortical tubers are present at birth. However, prenatal treatment would obviously increase the risk of serious side effects, including impairment of critical developmental processes that may be mTOR-dependent.
Prenatal treatment trials in mice have demonstrated potential side effects and incomplete rescue of the neuronal phenotype. In one mouse model, a single prenatal dose of rapamycin followed by postnatal continuation of rapamycin rescued the lethal phenotype of the mutant mice but they ultimately still developed an abnormal neurological phenotype including enlarged brains and cognitive deficits [58]. Further, in comparison with controls, reduced neonatal weight gain was seen. Difficulty with weight gain during prolonged postnatal treatment with rapamycin has also been seen in other mouse models [22•, 23, 28, 29, 36]. Limited data on safety of prenatal exposure to rapamycin in humans largely stem from transplant patients on rapamycin for immune suppression [59–61]. In these handful of case reports and seven women reported to the National Transplantation Pregnancy Registry, rapamycin does not appear to be associated with any specific birth defects although 3 of 7 pregnancies resulted in first trimester spontaneous abortions. Clearly more preclinical and clinical data are needed before considering prenatal treatment of rapamycin in TSC patients.
Besides prenatal treatment, an intermediate, less risky approach might involve identifying high-risk infants/children with TSC. Biomarkers for epilepsy have not been definitively established, but there is the possibility that specific EEG or brain MRI abnormalities may predict TSC patients with a worse neurologic prognosis and thus identify those patients most appropriate for antiepileptogenic therapy. For example, an abnormal EEG has been used to identify TSC patients prior to the onset of clinical seizures for starting vigabatrin treatment, which may decrease epilepsy severity and developmental delay [62]. If EEG is validated as an early biomarker for intractable epilepsy, a similar approach could be used with mTOR inhibitors. Additionally, although it seems likely that prolonged treatment with rapamycin may be necessary to sustain its effects, the optimal course of this treatment is yet to be determined. There is hope from mouse studies that intermittent treatment may both maintain an antiepileptic effect and reduce side effects [35•]. Beyond TSC, other high-risk populations, such as patients with TBI, could be considered for antiepileptogenic or neuroprotective trials, but should await more definitive data from animal models. Clearly the safety and efficacy of rapamycin as an antiepileptogenic treatment remain to be seen but in certain high-risk populations the time to consider formal trials is near.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–56.
McDaniel SS, Wong M. Therapeutic role of mammalian target of rapamycin (mTOR) inhibition in preventing epileptogenesis. Neurosci Lett. 2011;497:231–9.
Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:27–36.
Cho CH. Frontier of epilepsy research—mTOR signaling pathway. Exp Mol Med. 2011;43:231–74.
Weber JD, Gutmann DH. Deconvoluting mTOR biology. Cell Cycle. 2012;11:236–48.
Freeman JM, Kossoff EH. Ketosis and the ketogenic diet 2010: advances in treating epilepsy and other disorders. Adv Pediatr. 2010;57:315–29.
McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52:e7–e11.
Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, Fenning RS, Strauss K, Crino PB. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–602.
Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19:716–25.
• Krueger DA, Care MM, Holland K, Agricola K, et al. Everolimus for subependymal giant-cell astrocytomas in Tuberous Sclerosis. N Engl J Med. 2010;363:1801–11. This clinical trial showed that an mTOR inhibitor reduced the growth of subependymal giant cell astrocytomas in TSC patients, leading to FDA approval for this indication, and also provided some initial clinical data suggesting an effect of everolimus on seizures as a secondary measure.
Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G, Aronica E, Crino PB. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–87.
Schick V, Majores M, Engels G, Hartmann W, Elger CE, Schramm J, Schoch S, Becker AJ. Differential Pi3K-pathway activation in cortical tubers and focal cortical dysplasias with balloon cells. Brain Pathol. 2007;17:165–73.
Becker AJ, Urbach H, Scheffler BJ, Baden T, Normann S, Lahl R, Pennek HW, Tuxhorn I, Elger CE, Schramm J, Wiestler OD, Blumcke I. Focal cortical dysplasia of Taylor’s balloon cell type: mutational analysis of the TSC1 gene indicates a pathogenic relationship to Tuberous Sclerosis. Ann Neurol. 2002;52:29–37.
Schonberger A, Niehusmann P, Urbach H, Majores M, Grote A, Holthausen H, Blumcke I, Deckert M, Becker AJ. Increased frequency of distinct TSC2 alleleic variants in focal cortical dysplasias with balloon cells and mineralization. Neuropathology. 2009;29:559–65.
Gumbinger C, Rohsbach CB, Schulze-Bonhage A, Korinthenberg R, Zentner J, Haffner M, Fauser S. Focal cortical dysplasia: a genotype-phenotype analysis of polymorphisms and mutations in the TSC genes. Epilepsia. 2009;50:1396–408.
Grajkowski W, Kotulska K, Matyja E, Larysz-Brysz M, Mandera M, Roszkowski M, Domanska-Pakilela D, Lewik-Kowalik J, Jozwiak S. Expression of tuberin and hamartin in tuberous sclerosis complex-associated and sporadic cortical dysplasia of Taylor’s balloon cell type. Folia Neuropathol. 2008;46:43–8.
Lugnier C, Majores M, Fassunke J, Pernhorst K, Niehusmann P, Simon M, Nellist M, Schoch S, Becker A. Hamartin variants that are frequent in focal dysplasias and cortical tubers have reduced tuberin binding and aberrant subcellular distribution in vitro. J Neuropathol Exp Neurol. 2009;68:1136–46.
Ljungberg MC, Bhattacharjee MB, Lu Y, Armstrong DL, Yoshor D, Swann JW, Sheldon M, D’Arcangelo G. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol. 2006;60:420–9.
Miyata H, Chiang ACY, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56:510–9.
Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada KA, Gutmann DH. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–96.
Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–58.
• Goto J, Talos DM, Klein P, Qin W, Chekaluk YI, Anderl S, Malinowska IA, Di Nardo A, Bronson RT, Chan JA, Vinters HV, Kernie SG, Jensen FE, Sahin M, Kwiatkowski DJ. Regulable neural progenitor-specific TSC1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. PNAS. 2011;108:1070–9. This study described a novel mouse model of TSC and, consistent with previous studies, demonstrated that early postnatal treatment could prevent epilepsy and associated pathological abnormalities in these mice.
Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in TSC1—deficient forebrain and partial rescue by rapamycin. Neurobiol Dis. 2012;45:369–80.
Fu C, Cawthon B, Clinkscales W, Bruce A, Winzenburger P, Ess KC. GABAergic interneuron development and function is modulated by the Tsc1 gene. Cereb Cortex 2011 Oct 20 Epub ahead of print
Way SW, McKenna 3rd J, Mietzsch U, Reith RM, Wu HC, Gambello MJ. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum Mol Genet. 2009;18:1252–65.
Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a TSC2 +/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–8.
Zeng L, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. TSC2 gene inactivation causes a more severe epilepsy phenotype than TSC1 inactivation in a mouse model of Tuberous Sclerosis Complex. Hum Mol Genet. 2011;20:445–54.
Zeng L, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of Tuberous Sclerosis Complex. Ann Neurol. 2008;63:444–53.
Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski. Response of a neuronal model of Tuberous Sclerosis to mammalian target of Rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–32.
Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, et al. PTEN mutation spectrum and genotype - phenotype correlations in Bannayan–Riley–Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461–72.
Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia, and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403.
Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–11.
Kwon C, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci USA. 2003;100:12923–8.
Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D’Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2:389–98.
• Sunnen CN, Brewster AL, Lugo JN, Vanegas F, Turclos E, Mukhi S, Parghi D, D’Arcangelo G, Anderson AE. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia. 2011;52:2065–75. This study demonstrated that intermittent (2 weeks on, 4 weeks off) treatment with rapamycin prevented epilepsy progression and reduced side effects on growth in a genetic model of epilepsy.
Zhou J, Blundell J, Ogawa S, Kwon C, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–83.
• Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–72. This study provided evidence that mTOR inhibitors may inhibit epilepsy development in an animal model of acquired epilepsy following status epilepticus.
Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci. 2009;29:8259–69.
Huang X, Zhang H, Yang J, Wu J, McMahon J, Lin Y, Cao Z, Gruenthal M, Huang Y. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40:193–9.
• Buckmaster PS, Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci. 2011;31:2337–47. This study demonstrated that rapamycin inhibits mossy fiber sprouting, but did not prevent epilepsy in a model of acquired temporal lobe epilepsy.
Sliwa A, Plucinska G, Bednarczyk J, Lukasiuk K. Post-treatment with rapamycin does not prevent epileptogenesis in the amygdala stimulation model of temporal lobe epilepsy. Neurosci Lett. 2012; Epub ahead of print
Wong M. Rapamycin for treatment of epilepsy: antiseizure, antiepileptogenic, both, or neither? Epilepsy Curr. 2011;11:66–8.
Temkin NR. Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta-analysis of controlled trials. Epilepsia. 2001;42:515–24.
Chen S, Atkins CM, Liu CL, Alonso OF, Dietrich WD, Hu BR. Alterations in mammalian target of rapamycin signaling pathways after traumatic brain injury. J Cereb Blood Flow Metab. 2007;27:939–49.
Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93.
Guo D, Zeng LH, Brody DL, Wong M. mTOR inhibition has potential antiepileptogenic effects in a controlled cortical impact model of traumatic brain injury. Epilepsy Curr. 2011;11(Suppl 1): Abstract #1.014
Park J, Zhang J, Qiu J, Zhu X, Degterev A, Lo EH, Whalen MJ. Combination therapy targeting Akt and mammalian target of rapamycin improves functional outcome after controlled cortical impact. J Cereb Blood Flow Metab. 2012;32:330–40.
Berdichevsky Y, Saponjian Y, Mail M, Staley KJ. Organotypic culture model of post-traumatic epileptogenesis used as a medium-throughput screen of antiepileptic drugs. Epilepsy Curr. 2012;12(Suppl 1): Abstract #3.026
• Raffo E, Coppola A, Ono T, Briggs SW, Galanopoulo AS. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis. 2011;43:322–8. This study found that rapamycin can suppress spasms and reduce cognitive deficits in an animal model of infantile spasms.
Chachua T, Yum MS, Veliskova J, Velisek L. Validation of the rat model of cryptogenic infantile spasms. Epilepsia. 2011;52:1666–77.
Talos DM, Sun H, Jackson M, Joseph A, Fitzgerald E, Jensen F. Rapamycin attenuates the increases in seizure susceptibility and neuronal excitability following neonatal seizures in rat. Epilepsy Curr. 2011;11(Suppl 1):Abstract A.08
Sankar R, Auvin S, Kwon YS, Pineda E, Shin D, Mazarati A. Evaluation of development-specific targets for antiepileptogenic therapy using rapid kindling. Epilepsia. 2010;51(Suppl3):39–42.
Muncy J, Butler IJ, Koenig M. Rapaymycin reduces seizure frequency in Tuberous Sclerosis Complex. J Child Neurol. 2009;24:477.
Wilfong A, Krueger DA, Holland-Bouley K, Anderson AE, Agricola K, Schultz RJ, Tudor C, Mayws M, Lopez CM, Franz DN. Everolimus improves seizure control in tuberous sclerosis complex. American Epilepsy Society Meeting Abstracts 2011; Late-breaking Abstract #3.329
Daoud D, Scheld HH, Speckmann EJ, Gorji A. Rapamycin: brain excitability studied in vitro. Epilepsia. 2007;48:834–6.
Ruegg S, Baybis M, Juul H, Dichter M, Crino PB. Effects of rapamycin on gene expression, morphology, and electrophysiological properties of rat hippocampal neurons. Epilepsy Res. 2007;77:85–92.
Galanopoulou A, Crino P, Wong M, Buckmaster P, Raffo E. Rapamycin: from tuberous sclerosis and beyond. Epilepsia. 2009;50 Suppl 11:309.
Anderl S, Freeland M, Kwiatkowski DJ, Goto J. Therapeutic value of prenatal rapamycin treatment in a mouse brain model of tuberous sclerosis complex. Hum Mol Genet. 2011;20:4597–604.
Guardia O, del Rial MC, Casadei D. Pregnancy under sirolimus-based immunosuppression. Transplantation. 2006;81:636.
Sifontis NM, Coscia LA, Constantinescu S, Lavelanet AF, Moritz MJ, Armenti VT. Pregnancy outcomes in solid organ transplant recipients with exposure to mycophenolate mofetil or sirolimus. Transplantation. 2006;82:1698–702.
Chu SH, Liu KL, Chiang YJ, Wang HH, Lai PC. Sirolimus used during pregnancy in a living related renal transplant recipient: a case report. Transplant Proc. 2008;40:2446–8.
Jozwiak S, Kotulsaka K, Domanska-Pakilea D, Lojszczyk B, Syczewska M, Chmielewski D, Dunin-Wasocwicz D, Kmiec T, Szymkiewicz-Dangel J, Kornacka M, Kawalec W, Kuczynski D, Borkowska J, Tomaszek K, Jurkiewicz E, Respondek-Liberska M. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15:424–31.
Acknowledgments
M. Wong receives funding from the National Institutes of Health (R01 NS056872) and the Washington University/Pfizer Biomedical Research Collaboration.
Disclosure
Conflicts of interest: R.C.C. Ryther: has received travel/accommodations/meeting expenses unrelated to activities listed from the American Epilepsy Society; M. Wong: none.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ryther, R.C.C., Wong, M. Mammalian Target of Rapamycin (mTOR) Inhibition: Potential for Antiseizure, Antiepileptogenic, and Epileptostatic Therapy. Curr Neurol Neurosci Rep 12, 410–418 (2012). https://doi.org/10.1007/s11910-012-0276-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-012-0276-5