
Abstract
The oral sphingosine 1-phosphate receptor (S1PR) modulator fingolimod functionally antagonizes S1PR hereby blocking lymphocyte egress from secondary lymphoid organs to the peripheral blood circulation. This results in a reduction in peripheral lymphocyte counts, including potentially encephalitogenic T cells. In patients with relapsing multiple sclerosis fingolimod has been shown to be an effective treatment. In phase 2 and phase 3 studies fingolimod-treated patients had reduced disease activity clinically and in MRI. Although severe infectious complications occurred in single cases treated with fingolimod, the frequency of overall infections was comparable in fingolimod-treated patients and controls. Overall, in clinical studies fingolimod was well tolerated and had a favorable safety profile. In follow-up studies with continuous fingolimod, treatment showed sustained efficacy while being well tolerated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system (CNS) accompanied by substantial neurodegenerative processes. The heterogeneous clinical disease course, radiologic, and pathologic findings hamper a full understanding of the complex interrelationship of inflammatory response and tissue damage in different subgroups and phases of the disease [1, 2]. In recent years however, the immunologic mechanisms underlying the disease have been better understood and, based on this, new therapeutic approaches have been developed. This widened the therapeutic options substantially. Oral immunomodulators might broaden the therapeutic armamentarium and potentially provide effective, more convenient, and more selective treatment options of the inflammatory but also the neurodegenerative components of the disease. Among them, oral fingolimod is the first member of a new class of immunomodulators that targets sphingosine receptors. The drug has shown clinical and MRI efficacy in clinical phase 2 and 3 trials of relapsing MS and is now registered for the treatment of relapsing remitting MS in many countries [3••, 4••, 5••].
Mechanism of Action
Immunologic Effects
Fingolimod (FTY720) is a sphingosine analogue and is phosphorylated by sphingosine kinase 1 and 2 as its natural counterpart [6, 7]. Sphingosine-1 phosphate (S1P) binds to G-protein-coupled S1P receptors. To date, five subtypes of S1P receptors (S1P1–5) have been identified (Table 1) [8]. T cells, B cells, and natural killer (NK) cells mainly express S1P1 and to a smaller extent S1P3–5 (T cells), S1P3 (B cells), and S1P5 NK cells [9]. In the CNS, astrocytes express S1P1–3 and S1P5 [10], microglia cells express S1P1–3 and S1P5 [11, 12], and mature oligodendrocytes express S1P1, S1P3, and S1P5 [9, 13, 14]. In immune cells that recirculate to secondary lymphoid organs (SLOs) such as lymph nodes, binding of S1P to S1P1 mediates egress from SLOs [15]. Phosphorylated fingolimod binds as an analogue of S1P to the S1P receptors S1P1, S1P3, S1P4, and S1P5. Binding of phosphorylated fingolimod to S1P1 on lymphocytes results in internalization and degradation of the receptor [16]. As a result, S1P1-mediated egress of lymphocytes from SLOs to the periphery is blocked [17, 18]. Accordingly, inhibition of lymphocyte egress from SLOs by fingolimod pertains most likely also autoreactive T cells that mediate inflammation in MS lesions [19]. In fingolimod-treated patients with MS the percentages of CD4+ and CD8+ T lymphocytes are reduced [20]. Among them naïve T cells and central memory T cells (TCM) that recirculate to SLOs on a regular basis are predominantly reduced, whereas effector memory T cells (TEM) that do not recirculate on a regular basis to SLOs account for the majority of blood T lymphocytes in fingolimod-treated MS patients.
In recent years a T-cell subset characterized by the production of interleukin (IL)-17 has been demonstrated to play a central role in various models of human autoimmune such disease as experimental autoimmune encephalomyelitis, the animal model of MS [21–23]. In patients with MS the number of IL-17A mRNA-positive Th17 cells is elevated in peripheral blood and Th17 cells are enriched in the cerebrospinal fluid of patients with MS [22–24]. In MS lesions, Th17 cells are detected in high numbers and have an enhanced capacity to kill human neuronal cells when compared with control T cells [25, 26]. Blood CD4+ T cells with phenotypic and functional characteristics of Th17 cells are contained predominantly within the TCM subset, but are not found in TEM cells [27]. As a consequence, reduction of TCM by fingolimod treatment coincides with a major reduction of CD4+ Th17-like cells in blood. This finding was supported by the reduced expression of the Th17-specific transcription factor RORC2 and of barely detectable secretion of IL-17 by T cells of fingolimod-treated patients. Together these findings suggest that TEM and TEMRA cells are the predominant T-cell subtypes that remain in the peripheral blood of patients treated with fingolimod, and that egress from SLOs of naïve T cells, TCM, and Th17 cells are inhibited by fingolimod.
Besides T cells, numerous immune cells such as B cells, dendritic cells, and macrophages express sphingosine receptors [28]. Results from human studies and animal models indicate that functional antagonism of S1P inhibits also their egress from SLOs or migration into peripheral tissue [29–31], but this has not been reported with regard to fingolimod therapy in patients with MS. It was shown that the production of the proinflammatory cytokine IL-12p70 by blood-derived myeloid cells was inhibited [32]. In dendritic cells the production of the anti-inflammatory cytokine IL-10 was reduced, indicating together with the aforementioned reduction of IL-12 secretion additive anti-inflammatory effects.
CNS Effects of Fingolimod
In addition to its effects on lymphocytes, fingolimod has been reported to exert direct effects on CNS cells involved in the pathogenesis of MS such as astrocytes, oligodendrocytes, and microglia. Fingolimod and the phosphorylated active form (fingolimod-P) cross the blood–brain barrier and drug levels are found in steady-state conditions in higher concentrations in the brain parenchyma than in blood in treated animals [33]. In the CNS fingolimod colocalizes with myelin, presumably as a result of its lipophilic nature [34, 35]. In CNS tissue from patients with MS, mRNA levels of S1P receptors are upregulated in cells adjacent to MS lesions, and a strong increase in S1P1&3 expression on reactive astrocytes is found in active and chronic inactive MS lesions [36]. In rodents, progenitor and mature oligodendrocytes express S1P1 and S1P5 in vivo and in vitro [13].
Fingolimod induces modulation of S1P receptors on human oligodendrocyte progenitor cells (OPCs) and regulates differentiation of OPCs into oligodendrocytes [37–39]. In a system of lysolecithin-induced demyelination in organotypic cerebellar slice cultures fingolimod enhanced remyelination and process extension by OPCs and mature oligodendrocytes [40]. These effects were mediated primarily through S1P3 and S1P5. Fingolimod further rescued human mature myelin-producing oligodendrocytes from serum and glucose deprivation–induced apoptosis [41]. In astrocytes S1P and fingolimod augmented extracellular signal–regulated kinase (ERK), which mediates signaling for cell proliferation [42]. In the presence of tumor necrosis factor-α, S1P1 and S1P3 are upregulated in primary cultures of human astrocytes and fingolimod reduced the secretion of proinflammatory cytokines in these cells. This indicates regulation of S1P expression in astrocytes under proinflammatory conditions and direct anti-inflammatory effects of fingolimod in astrocytes [36]. In contrast to human astrocytes, fingolimod reduces ERK phosphorylation in human microglia cells [32].
Taken together, these findings implicate that fingolimod treatment, in addition to its effects on the composition of peripheral blood T-cell subsets, also has direct effects on oligodendrocytes, astrocytes, and microglia. These direct or indirect neuroprotective effects could serve as an explanation for the reduced loss of brain volume observed in fingolimod-treated patients [4••].
Clinical Outcomes
Clinical efficacy and safety of fingolimod in relapsing MS have been evaluated in one phase 2 and two phase 3 studies and resulted in approval of the drug for relapsing MS in 2010 by the US Food and Drug Administration (FDA). In the phase 2 study, 281 patients with active relapsing MS—defined by at least two relapses during the previous 2 years or one relapse in the previous year or one gadolinium-enhancing (Gd+) lesion on MRI—and an Expanded Disability Status Scale (EDSS) of 0 to 6.0 were equally randomized to receive oral fingolimod 1.25 mg or 5 mg or placebo once daily [3••]. The primary end point was the cumulative number of Gd+ lesions per patient in monthly MRIs. Both dosages of fingolimod reduced the total number of Gd+ lesions compared with placebo significantly. Also, several secondary end points were met: 1) The proportion of patients who were free from Gd+ lesions was greater in both fingolimod groups. 2) The annualized relapse rate (ARR) was reduced by 53% to 55% in fingolimod-treated patients. 3) Significantly more fingolimod-treated patients (86%) remained relapse-free when compared with the placebo group (66%). After completion of the core study patients were offered to enter an open-label, active-drug extension study in which the previously placebo-treated patients were randomized to fingolimod. Also, this group showed a significant decrease in the number of Gd+ lesions and the ARR after initiation of treatment with fingolimod.
In an open-label extension study following the phase 2 core study the reduced number of Gd+ lesions, which was observed during the first 6 months of fingolimod treatment in the core study, was sustained after 24 and 36 months and the majority of patients remained free from Gd+ lesions or new T2 lesions [43, 44]. In line with this most patients remained free from relapses and EDSS scores were stable. During months 15 to 24, patients receiving fingolimod 5 mg were switched to fingolimod 1.25 mg, which was related to fewer side effects while being comparably efficacious. MRI and clinical outcomes demonstrated a continuous beneficial effect of fingolimod treatment and confirmed the 24-month findings of the extension study.
The results of the phase 2 study were confirmed in two phase 3 studies. The FREEDOMS (FTY720 Research Evaluation Effects of Daily Oral therapy in MS) study was placebo-controlled; the TRANSFORMS (Trial Assessing Injectable Interferon Versus FTY720 Oral in RRMS) study compared fingolimod to the established MS therapeutic interferon-β1a (IFN-β1a; intramuscular once weekly).
In the FREEDOMS study patients were randomized to receive oral fingolimod 0.5 mg, fingolimod 1.25 mg, or placebo once daily over 24 months [4••]. A total of 1272 patients with relapsing MS with an EDSS between 0 and 5.5 who had one relapse during the last year or two relapses during the previous 2 years were recruited for the study. Mean age of patients was 37 years. Approximately 60% were treatment-naïve, mean disease duration was 8 years, and the mean EDSS was 2.4. Compared with placebo, treatment with fingolimod reduced the ARR significantly (fingolimod 0.5 mg: 0.18; fingolimod 1.25 mg: 0.16; placebo 0.4), which corresponds to a relative risk reduction of 54% to 60%. Also, the time to confirmed disability progression after 3 and 6 months was prolonged significantly in both fingolimod groups. In addition, several secondary end points were favorable for fingolimod: Fingolimod treatment decreased the number of Gd+ lesions and the number of new or enlarged T2 lesions significantly. Increase of the volume of T1-hypointense lesions as a marker for tissue damage and whole-brain atrophy were also reduced by fingolimod.
In the TRANSFORMS study patients were randomized in a double-dummy design to oral fingolimod 0.5 mg, fingolimod 1.25 mg, or IFN-β1a intramuscularly once weekly [5••]. A total of 1292 patients with relapsing MS with an EDSS between 0 and 5.5 who had one relapse during the last year or two relapses during the previous 2 years were recruited for the study. Mean age was 36 years, and the majority of patients (~56%) had previous treatment for MS, mostly IFN-β or glatiramer acetate. Average disease duration was 7.4 years, and the patients had a mean EDSS of 2.2. In both fingolimod groups a significant reduction of the ARR compared with IFN-β1a was noted (fingolimod 0.5 mg: 0.16; fingolimod 1.25 mg: 0.2; IFN-β1a: 0.33). Corresponding to this the proportion of relapse-free patients was significantly higher in fingolimod-treated patients (fingolimod 0.5 mg: 82.6%; fingolimod 1.25 mg: 79.8%; IFN-β1a: 69.3%). Patients in the fingolimod groups also had less Gd+ lesions, a reduced number of new or enlarged T2 lesions, and less brain atrophy.
In an extension of the TRANSFORMS study patients who were treated with IFN-β1a in the core study were randomly assigned to receive 0.5 mg or 1.25 mg of fingolimod, whereas the initially fingolimod-treated patients continued their treatment [45••]. Primary end points were the ARR, disability progression, and MRI outcomes. Patients receiving continuous fingolimod had unchanged benefits in the ARR. Patients who were switched from IFN-β1a to fingolimod showed a significant decrease of the ARR. Also, numbers of new or newly enlarging T2 lesions, Gd+ lesions, and the rate of brain volume decline were reduced. Compared with patients who had switched from IFN-β1a therapy to fingolimod, continuously fingolimod-treated patients had a significantly lower ARR and reduced inflammatory activity in brain MRI. Side effects shifted in patients who were switched to fingolimod to a profile that is known from other studies in fingolimod-treated patients with MS.
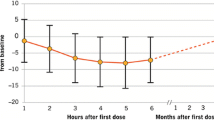
In all clinical studies fingolimod was well tolerated and had a favorable safety profile. Apart from the fingolimod 5-mg group, in the phase 2 study the frequency of adverse events in fingolimod-treated patients was comparable with controls. Therefore, patients treated with fingolimod 5 mg were switched to fingolimod 1.25 mg during months 15 to 24 of the phase 2 extension study [44]. The vast majority of adverse events were mild to moderate in all study groups. The most common serious adverse events in fingolimod-treated patients were transient bradycardia and atrioventricular block related to the first dose of the medication, MS relapses, and basal cell carcinoma. Macular edema occurred in 1% of the patients in the 1.25-mg group and 0.5% of the 0.5-mg group and resolved in most patients after discontinuation of fingolimod therapy. Other adverse events occurred in less than 1% of the study populations.
In all clinical studies the frequency of overall infections was comparable in fingolimod-treated patients and controls. Nasopharyngitis was the most frequent infection with no differences between patients treated with fingolimod 0.5 mg or 1.25 mg and controls. In the placebo-controlled phase 3 study, lower respiratory tract infections (including bronchitis and pneumonia) were more frequent in patients treated with fingolimod. Two deaths related to herpes virus infections occurred during the TRANSFORMS study. One patient developed primary disseminated varicella zoster infection after exposure to a child with chicken pox while under corticosteroid treatment. One patient developed lethal herpes simplex encephalitis, primarily treated with intravenous corticosteroids for suspected relapse of MS.
Besides lymphopenia, elevated alanine aminotransferase (ALAT) levels were common abnormal laboratory findings in all study groups, but were more frequent in patients treated with fingolimod. Up to 12.5% of patients had ALAT levels three times the upper limit of the normal range or more. In all patients liver enzyme abnormalities returned to normal after drug discontinuation.
After completing the core phase 3 studies, patients were offered to enter into open-label, uncontrolled active-drug extension phases. Patients previously treated with placebo or IFN-β1a were switched to fingolimod 0.5 mg or 1.25 mg, whereas the others continue their assigned dosages.
Currently, all patients from the phase 2 and the two phase 3 studies are monitored in a single extension study. Herein all patients were switched to fingolimod 0.5 mg. Patients are scheduled every third month for clinical and laboratory assessments. The study aims at compiling additional data on the long-term safety and tolerability of fingolimod therapy.
Future Directions
After introduction of fingolimod for the treatment of relapsing MS, a phase 4 clinical program aims to assess tolerability and safety of the drug when used in patients under non-study condition. In addition to the follow-up studies of the clinical studies, this will also help to assess long-term tolerability of fingolimod. In autumn 2011, results of two phase 3b studies of fingolimod in MS will be available. One study assessed the effect of fingolimod treatment on the induction of an antigen-specific immune response to influenza vaccination as a model for a newly induced immune response and on the induction of a recall immune response to tetanus booster vaccination. In a second phase 3b study, tolerability of initiation of fingolimod treatment without the currently required 6-hour observation period for cardiovascular monitoring after the first dose was assessed. Because the mode of action may also have direct neuroprotective effects, fingolimod is being evaluated in a phase 3 study in patients with primary progressive MS. Ongoing phase 2 studies are evaluating the clinical efficacy and safety of new compounds that are more specific for certain S1P receptors with the aim to further improve the efficacy/safety profile.
Conclusions
Oral fingolimod is the first member of the new class of S1P1 modulators and has been approved by the FDA and European Medicines Agency for the treatment of relapsing MS. Two large phase 3 clinical trials showed a superior efficacy of fingolimod compared to both placebo and IFN-β1a intramuscularly. Fingolimod was well tolerated and safe. The overall incidence of infections, including severe and serious infections, was comparable between control groups and those receiving fingolimod 0.5 mg. However, a slightly increased incidence of lower respiratory tract and lung infections (mainly bronchitis) was seen in fingolimod-treated patients. Follow-up studies will continue to assess the long-term tolerability and safety of fingolimod. The study addressing the effects of fingolimod in primary progressive MS will potentially help to better characterize the neuroprotective effect of fingolimod.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Kappos L, Moeri D, Radue EW, et al. Predictive value of gadolinium-enhanced magnetic resonance imaging for relapse rate and changes in disability or impairment in multiple sclerosis: a meta-analysis. Gadolinium MRI meta-analysis group. Lancet. 1999;353:964–9.
Bar-Or A. The immunology of multiple sclerosis. Semin Neurol. 2008;28:29–45.
•• Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–40. The first POC study with fingolimod in Multiple sclerosis.
•• Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. This phase 3 study confirmed the safety and tolerability and also the efficacy of oral fingolimod in patients with relapsing MS.
•• Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–15. This phase 3 study confirmed the safety and tolerability of oral fingolimod in patients with relapsing MS and also demonstrated increased efficacy when compared with IFN-β1a.
Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–7.
Paugh SW, Payne SG, Barbour SE, et al. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003;554:189–93.
Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem. 2009;78:743–68.
Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol. 2005;5:560–70.
Rao TS, Lariosa-Willingham KD, Lin FF, et al. Pharmacological characterization of lysophospholipid receptor signal transduction pathways in rat cerebrocortical astrocytes. Brain Res. 2003;990:182–94.
Harada J, Foley M, Moskowitz MA, Waeber C. Sphingosine-1-phosphate induces proliferation and morphological changes of neural progenitor cells. J Neurochem. 2004;88:1026–39.
Tham CS, Lin FF, Rao TS, et al. Microglial activation state and lysophospholipid acid receptor expression. Int J Dev Neurosci. 2003;21:431–43.
Yu N, Lariosa-Willingham KD, Lin FF, et al. Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia. 2004;45:17–27.
Chun J, Weiner JA, Fukushima N, et al. Neurobiology of receptor-mediated lysophospholipid signaling. From the first lysophospholipid receptor to roles in nervous system function and development. Ann N Y Acad Sci. 2000;905:110–7.
Mandala S, Hajdu R, Bergstrom J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–9.
Graler MH, Goetzl EJ. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004;18:551–3.
Chiba K, Yanagawa Y, Masubuchi Y, et al. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J Immunol. 1998;160:5037–44.
Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–60.
Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33:91–101.
Mehling M, Brinkmann V, Antel J, et al. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology. 2008;71:1261–7.
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517.
Reboldi A, Coisne C, Baumjohann D, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–23.
Brucklacher-Waldert V, Stuerner K, Kolster M, et al. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain. 2009;132:3329–41.
Matusevicius D, Kivisakk P, He B, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–4.
Tzartos JS, Friese MA, Craner MJ, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55.
Kebir H, Kreymborg K, Ifergan I, et al. Human T(H)17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–5.
Mehling M, Lindberg R, Raulf F, et al. Th17 central memory T cells are reduced by FTY720 in patients with multiple sclerosis. Neurology. 2010;75:403–10.
Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115:84–105.
Bohler T, Schutz M, Budde K, et al. Differential effects of single dose FTY720 on CD62L+B-cells in stable renal allograft recipients. Int Immunopharmacol. 2007;7:88–95.
Vaessen LM, van Besouw NM, Mol WM, et al. FTY720 treatment of kidney transplant patients: a differential effect on B cells, naive T cells, memory T cells and NK cells. Transpl Immunol. 2006;15:281–8.
Lan YY, De Creus A, Colvin BL, et al. The sphingosine-1-phosphate receptor agonist FTY720 modulates dendritic cell trafficking in vivo. Am J Transplant. 2005;5:2649–59.
Durafourt BA, Lambert C, Johnson TA, et al. Differential responses of human microglia and blood-derived myeloid cells to FTY720. J Neuroimmunol. 2011;230:10–6.
Sanchez T, Hla T. Structural and functional characteristics of S1P receptors. J Cell Biochem. 2004;92:913–22.
Li H, Meno-Tetang GM, Chiba K, et al. Pharmacokinetics and cell trafficking dynamics of 2-amino-2-[2-(4-octylphenyl)ethyl]propane-1,3-diol hydrochloride (FTY720) in cynomolgus monkeys after single oral and intravenous doses. J Pharmacol Exp Ther. 2002;301:519–26.
Meno-Tetang GM, Li H, Mis S, et al. Physiologically based pharmacokinetic modeling of FTY720 (2-amino-2[2-(−4-octylphenyl)ethyl]propane-1,3-diol hydrochloride) in rats after oral and intravenous doses. Drug Metab Dispos. 2006;34:1480–7.
Van Doorn R, Van Horssen J, Verzijl D, et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia. 2010;58:1465–76.
Miron VE, Jung CG, Kim HJ, et al. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol. 2008;63:61–71.
Coelho RP, Payne SG, Bittman R, et al. The immunomodulator FTY720 has a direct cytoprotective effect in oligodendrocyte progenitors. J Pharmacol Exp Ther. 2007;323:626–35.
Jung CG, Kim HJ, Miron VE, et al. Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia. 2007;55:1656–67.
Miron VE, Ludwin SK, Darlington PJ, et al. Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am J Pathol. 2010;176:2682–94.
Miron VE, Hall JA, Kennedy TE, et al. Cyclical and dose-dependent responses of adult human mature oligodendrocytes to fingolimod. Am J Pathol. 2008;173:1143–52.
Osinde M, Mullershausen F, Dev KK. Phosphorylated FTY720 stimulates ERK phosphorylation in astrocytes via S1P receptors. Neuropharmacology. 2007;52:1210–8.
O’Connor P, Comi G, Montalban X, et al. Oral fingolimod (FTY720) in multiple sclerosis: two-year results of a phase II extension study. Neurology. 2009;72:73–9.
Comi G, O’Connor P, Montalban X, et al. Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3-year results. Mult Scler. 2010;16:197–207.
•• Khatri B, Barkhof F, Comi G, et al. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: a randomised extension of the TRANSFORMS study. Lancet Neurol. 2011;10:520–9. This extension study from the phase 3 TRANSFORMS study demonstrated that patients receiving continuous fingolimod had unchanged beneficial effects, whereas patients who were switched from IFN-β1a to fingolimod showed a significant decrease of clinical and MRI disease activity.
Disclosure
Conflicts of interest: M. Mehling: none; L. Kappos: has received grant support to the University Hospital for his participation as principal investigator, member, or chair of planning and steering committees or advisory boards in corporate-sponsored clinical trials in multiple sclerosis and other neurological diseases sponsored by the following companies: Actelion, Advancell, Allozyne, BaroFold, Bayer Health Care Pharmaceuticals, Bayer Schering Pharma, Bayhill, Biogen Idec, BioMarin, CLC Behring, Elan, Genmab, GeNeuro SA, Genmark, GlaxoSmithKline, Eli Lilly, Merck Serono, MediciNova, Novartis, Novonordisk, Pepimmune, Sanofi-Aventis, Santhera, Roche, Teva, UCB, and Wyeth; research and clinical operations of the MS Center in Basel have been supported by nonrestricted grants from one or more of these companies, and by grants from the Swiss MS Society, the Swiss National Research Foundation, the European Union, the Gianni Rubatto, Novartis and Roche Research Foundations; T. Derfuss: has been a consultant for Novartis, Merck Serono, Biogen Idec, and Bayer Schering; has received grant support from Novartis and Merck Serono; and has received travel/accommodations expenses covered or reimbursed from Novartis, Biogen Idec, Merck Serono, and Bayer Schering.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mehling, M., Kappos, L. & Derfuss, T. Fingolimod for Multiple Sclerosis: Mechanism of Action, Clinical Outcomes, and Future Directions. Curr Neurol Neurosci Rep 11, 492–497 (2011). https://doi.org/10.1007/s11910-011-0216-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-011-0216-9