
Abstract
Purpose of Review
To highlight the role of the brain melanocortin 4 receptor (MC4R) for sympathetic nervous system (SNS) activation in hypertension.
Recent Findings
Hypertension is the most significant risk factor for developing cardiovascular disease. Although excess weight gain is associated with at least two thirds of primary hypertension cases, the pathophysiological mechanisms involved remain the subject of intense investigation. Multiple studies demonstrate an important role for increased sympathetic nervous system (SNS) activity in development and maintenance of hypertension, and that the brain MC4R modulates SNS activity to thermogenic, cardiovascular, and kidney tissues. These studies also support the concept that MC4R activation is critical for obesity-induced hypertension as well as other forms of hypertension associated with increased SNS activity.
Summary
MC4R is a potential target for antiobesity therapy, although there are challenges in using MC4R agonists to induce weight loss without evoking increases in SNS activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases remain the leading cause of death in the USA and worldwide despite recent advances in diagnosis and treatment https://www.who.int/cardiovascular_diseases/world-heart-day/en/. Amongst the many causes of cardiovascular diseases, elevated blood pressure (BP) is one of the most critical risk factors for vascular injury, heart failure, stroke, and renal disease [1, 2]. Several mechanisms, including overactivation of the sympathetic nervous system (SNS), have been proposed to participate in development and maintenance of primary (essential) hypertension, the most prevalent form of hypertension [3•]. Excess adiposity is thought to be a major driver of SNS activity and may account for as much as 65–75% of the risk for developing primary hypertension. However, the mechanisms responsible for elevated SNS activity observed in obesity-induced hypertension as well as in many non-obese individuals with hypertension are still unclear.
Several factors have been proposed to explain increased SNS activity in hypertensive individuals, particularly in those who exhibit increased visceral adiposity [3•, 4•]. In this brief review, we focus on the importance of the brain melanocortin 4 receptor (MC4R) in modulating SNS activity and its contribution to elevated SNS in obesity-induced hypertension as well as in other forms of hypertension not accompanied by excess adiposity.
MC4R Regulates Body Weight Homeostasis by Modulating Appetite and Energy Expenditure
The MC4R is a G protein–coupled 7 transmembrane receptor that belongs to the melanocortin receptor family and it is thought to act as the dominant efferent arm of the brain melanocortin system’s actions on body weight regulation. The most important endogenous MC4R agonist is α-melanocyte-stimulating hormone (α-MSH), produced from the cleavage of proopiomelanocortin (POMC) protein in a specific set of neurons (POMC neurons) that, when stimulated, induce satiety [5••, 6••]. POMC neurons are located mainly in the arcuate nucleus of the hypothalamus and send projections to several nuclei where they release α-MSH to activate MC4R leading to reduced appetite and increased thermogenesis [5••, 6••]. It is estimated that impaired MC4R activation, caused by mutations of the MC4R or the POMC gene, accounts for as much as 5–6% of early-onset morbid obesity in humans [5••, 6••]. Humans and experimental animals with MC4R deficiency or loss-of-function mutations exhibit voracious appetite and reduced energy expenditure compared with obese controls with wild-type genotypes [5••, 6••].
Balthasar et al. showed that restoration of MC4R expression in the paraventricular nucleus of the hypothalamus (PVN) of MC4R-deficient mice decreased appetite to normal but did not substantially increase energy expenditure, resulting in only partial amelioration of the severe obesity observed in MC4R deficient mice [7]. do Carmo et al. showed that restoration of MC4R function specifically in POMC neurons also partially attenuated the obese phenotype of MC4R-deficient mice, mainly by restoring MC4R control of energy expenditure without diminishing the increased appetite of these mice [8]. These studies highlight the importance of MC4R in modulating appetite and energy expenditure to influence body weight regulation.
These and other observations documenting the importance of MC4R in body weight regulation led to the notion that MC4R may be a potential target for therapies to promote weight loss. However, as discussed below, MC4R not only regulates appetite and SNS activity to thermogenic tissues but also increases SNS activity to tissues and organs that are important for BP regulation.
MC4R Modulates SNS Activity and Plays a Major Role in Obesity-Associated Hypertension
Activation of brain MC4R by acute intracerebroventricular (ICV) injections of pharmacological agonists increases renal SNS activity and heart rate (HR) [9, 10]. Increased renal SNS activity promotes sodium retention and renin release, actions that if sustained can elevate BP. In fact, chronic ICV infusions of MC4R agonists cause sustained elevations in BP despite weight loss which normally tends to lower BP [11]. Similar observations are found in humans injected subcutaneously with MC4R agonists that cross the blood-brain barrier [12]. Perhaps the most compelling evidence that MC4R influences BP regulation comes from studies demonstrating that pharmacological blockade of MC4R or MC4R deficiency causes rapid severe weight gain and increased adiposity while reducing HR and SNS activity and prevents the rise in BP that usually accompanies obesity [13, 14]. Humans with loss-of-function MC4R mutations are morbidly obese and exhibit many characteristics of the metabolic syndrome, but the prevalence of hypertension in these subjects is lower than expected when compared with obese individuals with normal MC4R function [5••, 6••, 12].
MC4R activation not only contributes to the elevated basal SNS activity in obese individuals, but modulates SNS to stress stimuli. Humans with MC4R deficiency show reduced SNS responses to various stimuli including acute apnea and waking [15]. Experimental animal models with MC4R deficiency recapitulate the phenotypes observed in humans with impaired MC4R function and exhibit normal BP despite morbid obesity and attenuated BP and HR responses to stress stimuli [8, 14]. MC4R-deficient mice are also completely unresponsive to the effects of leptin to increase renal SNS activity, BP, and HR [16]. Leptin, an adipocyte-derived peptide that is produced in proportion to the degree of adiposity, was shown to be a critical factor linking excess weight gain with increased SNS activity and elevated BP in obesity [17]. The fact that MC4R deficiency abolishes the effects of hyperleptinemia on sympathetic activity and BP suggests that MC4R activation may be required for hyperleptinemia and obesity to be associated with increased SNS activity and hypertension [18].
MC4R Contributes to BP Regulation in Non-obese Forms of Hypertension Associated with Sympathetic Activation
In addition to its unequivocal role in obesity-associated hypertension, MC4R also appears to contribute importantly to elevated BP in some other forms of hypertension not accompanied by obesity. For example, blockade of brain MC4R markedly attenuated hypertension induced by chronic administration of the nitric oxide synthase inhibitor, l-NAME [19]. Activation of brain MC4R using pharmacological agonists also exacerbated the hypertensive effects of l-NAME [20]. This is of particular importance given the fact that obesity is also normally associated with impaired nitric oxide availability which may potentiate the impact of MC4R activation on obesity-associated hypertension.
Another widely used experimental model of hypertension that exhibits elevated SNS activity is the spontaneously hypertensive rat (SHR). These rats are not obese but spontaneously develop elevated BP at an early age. In SHR, chronic antagonism of brain MC4R significantly reduced BP to levels comparable with those achieved by adrenergic receptor blockade [21] suggesting that basal MC4R activation in SHR plays an important role in maintaining elevated SNS activity and BP.
MC4R activation also appears to contribute to hypertension observed in an experimental model of pre-eclampsia induced by placental ischemia [22]; in pregnant MC4R heterozygous dams submitted to placental ischemia, the rise in BP was significantly attenuated compared with wild-type pregnant controls with placental ischemia [22]. In a fetal programming model of hypertension associated with increased SNS activity, alterations in the perinatal environment have been suggested to alter MC4R activity in the offspring. For instance, hyperleptinemia during the neonatal period leads to augmented BP later in life, an effect that is not observed in MC4R-deficient animals but that can be rescued in mice with selective restoration of MC4R function in PVN neurons [23].
Collectively, these studies demonstrate the importance of MC4R in regulating SNS activity and BP not only in obesity, the most common cause of primary hypertension, but also in other forms of hypertension associated with increased SNS activity. The notion that MC4R activation contributes to elevated BP, particularly when hypertension is associated with increased SNS activity, is also supported by studies showing a minimal impact of MC4R antagonism on BP when SNS activity is normal or reduced such as in lean non-hypertensive animals or in low-dose angiotensin II–induced hypertension [11, 19]. An exception to this general finding is the observation that MC4R deficiency or pharmacological antagonism did not attenuate hypertension induced by chronic intermittent hypoxia [24•], a model in which hypertension is thought to be driven, at least in part, by increased SNS activity [25]. This intriguing finding suggests that MC4R modulation of SNS activity and BP is complex and not yet fully understood.
Mechanisms of MC4R Control of SNS Activity and BP
As discussed above, obesity is a major cause of primary hypertension. Unfortunately, lifestyle modifications have provided limited benefits to a significant portion of obese subjects at risk for developing hypertension and other cardiometabolic diseases. Therefore, pharmacological approaches and bariatric surgery continue to receive attention as a means to reduce the incidence of obesity and alleviate its cardiometabolic consequences [26]. Because of its ability to reduce appetite and to increase energy expenditure, the MC4R is a potentially attractive target for the development of antiobesity drugs. However, enthusiasm for early generations of MC4R agonists was dampened by the adverse effects of MC4R activation on other physiological functions, including increases in SNS activity and BP [6••].
Recent studies, however, suggest that activation of MC4R in distinct areas of the brain and, perhaps, activation of specific intracellular messengers downstream of MC4R may differentially regulate SNS activity to various target tissues while preserving the beneficial effects of MC4R activation on food intake, glucose metabolism, and energy expenditure. Thus, a better understanding of the mechanisms by which MC4R regulates SNS activity may lead to the development of agonists that reduce appetite and increase thermogenesis without increasing BP due to increased SNS activity to the kidneys, heart, and blood vessels.
MC4Rs are expressed in several areas of the CNS. The regions with significant MC4R expression include the PVN, lateral hypothalamus, amygdala, dorsal motor complex containing the nucleus of the tractus solitarius (NTS) and the dorsal motor nucleus of the vagus (DMV) [5••, 7, 27, 28], and preganglionic sympathetic neurons of the intermediolateral medulla (IML) [27]. All of these nuclei participate in control of autonomic function. For example, acute activation of MC4R in PVN neurons raised renal SNS activity and BP [9] and MC4R blockade in these same neurons abolished insulin-induced elevation of lumbar SNS activity [29]. The observation that restoration of MC4R function in PVN neurons in whole-body MC4R–deficient mice did not restore normal energy expenditure in these mice [7] may suggest that these neurons contribute to MC4R’s modulation of renal SNS activity but not SNS activity to thermogenic tissues.
MC4Rs in brainstem nuclei also appear to play an important role in controlling SNS activity and BP. Using transgenic mice that express MC4R only in cholinergic preganglionic sympathetic neurons in the NTS/DMV and IML, Sohn et al. restored the obesity-associated increases in BP and sympathetic activation in response to MC4R agonists that were absent in MC4R-deficient mice [30]. In a subsequent study, the authors showed that MC4R in cholinergic preganglionic sympathetic neurons of the DMV/NTS and IML also contribute to the thermogenic effects of MC4R activation [31]. However, the specific neuronal populations by which MC4R differentially regulates appetite, metabolism, and cardiovascular function remain as an important area for future investigation.
Another important aspect of MC4R control of appetite, metabolic function, SNS activity, and BP is its intrinsic activity during the apparent absence of known ligands [5••, 6••, 28]. This may help explain why some effects of MC4R activation during chronic infusions of exogenous agonists are less pronounced when compared with the long-term effects of MC4R blockade [11, 32]. For instance, MC4R antagonism in normotensive animals evoked marked bradycardia while chronic MC4R activation using synthetic agonists caused modest increases in HR [11]. Although the impact of MC4R intrinsic activity in regulating sympathetic drive, HR, and BP is still poorly understood, it may also explain the important contribution of MC4R to the elevated SNS activity and BP observed in conditions where the POMC-α-MSH-MC4R axis does not appear to be overstimulated [19, 21].
The precise mechanisms that regulate MC4R intrinsic activity are still not fully understood but appear to involve interactions of MC4R with proteins that regulate MC4R endocytosis and trafficking from the Golgi apparatus and endoplasmic reticulum to the plasma membrane [5••, 6••] as well as MC4R interaction with accessory proteins [5••, 6••, 33]. Kay et al. showed increased MC4R intrinsic activity in HEK293 cells transfected with the accessory protein hMRAPα [33]. Some forms of obesity caused by reduced MC4R function appear to be due to intracellular retention of MC4R in the endoplasmic reticulum as misfolded and ubiquitinated protein; this retention can be ameliorated with the use of chemical chaperons that improve protein folding and inhibitors of ubiquitination that inhibit proteasome protein degradation [5••]. The importance of the proteins that regulate MC4R trafficking and intrinsic activity in contributing to the long-term physiological effects of MC4R on SNS activity and BP, however, remains unclear and requires additional investigation.
Recent studies also indicate that MC4R intracellular signaling pathways are more complex than the commonly accepted view that MC4R mainly triggers stimulatory G protein (Gs) leading to increased activation of adenylyl cyclase to convert ATP to cAMP to stimulate protein kinase A (PKA) activity to increase neuronal firing [6••]. For example, in hypothalamic GT1-1 cells, MC4R-induced stimulation of Gq/11 signaling via activation of phospholipase C led to increased intracellular Ca2+ levels [34], while disruption of MC4R-induced Gq/11 signaling in PVN neurons caused obesity and hyperphagia and abolished the anorexic effects of MTII, a synthetic MC4R agonist [35•]. It is also important to note that while deletion of Gq/11 in PVN neurons abolished the effects of MC4R agonist on food intake, it did not attenuate the effect of MC4R activation to increase BP and HR, as only mice with disrupted Gs signaling in PVN neurons were unresponsive to MTII’s effects to raise BP and HR [35•].
Litt et al. showed that stimulation of MC4R in hypothalamic slices regulates inward rectifying potassium (Kir7.1) channel activity to depolarize neurons via a Gs-independent mechanism [36]. Subsequently, the authors demonstrated that deletion of the gene encoding Kir7.1 channels specifically in MC4R expressing neurons caused late-onset obesity, glucose intolerance, augmented linear growth, and resistance to the effects of MC4R agonists to depolarize PVN neurons [37•]; however, some MC4R-mediated actions remained intact including AgRP-induced hyperphagia and MC4R stimulation of peptide YY release from intestinal L cells. These studies add complexity to MC4R-mediated regulation of body weight and cardiovascular function but highlight the potential therapeutic value of selectively triggering some of the various effects of MC4R activation.
Future Directions in the Development of MC4R Agonists
Progress on MC4R biology in the last 10 to 15 years has markedly enhanced our understanding of MC4R trafficking, intracellular pathways, and divergent control of its various metabolic and cardiovascular actions by different neuronal populations. This has renewed hope that novel MC4R agonists can be developed to elicit MC4R-mediated beneficial effects but not its undesirable ones such as SNS stimulation and increased BP; thus, in a best-case scenario, an MC4R agonist might be developed that could induce satiety and increase energy expenditure to promote weight loss and improve the metabolic profile without increasing SNS activity to the heart, blood vessels, and kidneys and elevating BP. Several peptides and non-peptide agonists of MC4R have been tested in experimental animals and humans with leptin-POMC-MC4R axis mutations as well as in obese individuals with normal genotypes (reviewed in [5••, 6••]). Unfortunately, most of these agonists have elicited sympathetic activation and elevated BP, and other undesirable effects. A few of these agonists, however, appear to promote weight loss with minimal impact on BP. For instance, Chen et al. showed increased resting energy expenditure in 12 obese volunteers after a 72-h treatment with the MC4R agonist setmelanotide, also known as RM-493, with no significant increases in BP [38]. Kuhnen et al. observed an average 15% decrease in body weight after 3 months of treatment with setmelanotide in two patients with POMC deficiency with no increase in BP and perhaps even a decrease in BP following the weight loss [39••]. In a longer study in three patients with leptin receptor deficiency, the authors showed substantial weight loss and reduced hunger with setmelanotide treatment and, as with previous studies, no adverse cardiovascular effects were noted [40••]. Whether setmelanotide can be used in obese subjects who do not have leptin-POMC mutations for long periods of time with no adverse SNS and BP effects is still unclear.
Other beneficial effects of MC4R activation have been reported. For example, Liu and colleagues observed that MC4R activation using the agonist RO27-3225 significantly protected against brain injury, including reduced brain edema, blood-brain barrier permeability, and inflammation of cerebral tissues, evoked by intra-abdominal hypertension and hemorrhagic shock [41]. Gava et al. also showed improved cardiac function in a model of heart failure induced by permanent ligation of the left anterior descending coronary artery [42], whereas loss of MC4R function has been associated with dilated cardiomyopathy in mice [43]. These studies along with MC4R’s role in appetite, body weight homeostasis, SNS activity, and BP reinforce the importance of understanding how the MC4R exerts its multiple actions and how these effects can be differentially activated by synthetic agonists.
Conclusions
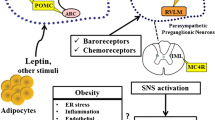
Hypertension is the most significant risk factor for cardiovascular diseases. Although the etiology of primary hypertension is still elusive, increased SNS activity contributes importantly to elevated BP in many patients with hypertension, especially when associated with obesity. The MC4R, a major regulator of appetite and body weight homeostasis, also plays a key role in modulating SNS activity. As summarized in Fig. 1, perinatal environment as well as obesity-dependent and obesity-independent factors later in life may be critical to the elevated SNS activity observed in several forms of hypertension. Recent promising studies suggest that various actions of MC4R activation can be selectively and independently stimulated according to the neuronal populations that are affected, by altering MC4R intrinsic activity, or by specific activation of MC4R’s intracellular pathways. Understanding the mechanisms by which MC4R differentially regulates metabolic and cardiovascular function is key to developing novel, more effective pharmacological approaches to treat obesity and other metabolic diseases with minimal adverse impact on SNS activity and BP regulation.

Summary of the impact of various factors on POMC neuronal firing and MC4R activation leading to increased SNS activity, renal sodium retention, and elevated blood pressure. MC4R melanocortin 4 receptor, NO nitric oxide, POMC proopiomelanocortin, SNS sympathetic nervous system
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Dai S, Huang B, Zou Y, Liu Y. Associations of dipping and non-dipping hypertension with cardiovascular diseases in patients with dyslipidemia. Arch Med Sci. 2019;15(2):337–42.
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365(9455):217–23.
• Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity, kidney dysfunction and hypertension: mechanistic links. Nature Rev Nephrol. 2019; (in press). A recent comprehensive review on the mechanisms linking obesity, renal dysfunction, and hypertension. This review highlights several potential mechanisms including compressive physical forces, hormonal changes, and the importance of the sympathetic nervous system in the development of obesity hypertension.
Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116(6):991–1006.
•• Baldini G, Phelan KD. The melanocortin pathway and control of appetite-progress and therapeutic implications. J Endocrinol. 2019;241(1):R1–R33. A recent review on basic aspects of MC4R structure, function, and physiological importance with focus on body weight regulation.
•• Kuhnen P, Krude H, Biebermann H. Melanocortin-4 receptor signalling: importance for weight regulation and obesity treatment. Trends Mol Med. 2019;25(2):136–48. A recent short review on MC4R structure and common mutations observed in humans, MC4R importance for appetite and body weight regulation, and recent advances in the development of pharmacological agonists.
Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505.
do Carmo JM, da Silva AA, Rushing JS, Pace B, Hall JE. Differential control of metabolic and cardiovascular functions by melanocortin-4 receptors in proopiomelanocortin neurons. Am J Physiol Regul Integr Comp Physiol. 2013;305(4):R359–68.
Li P, Cui BP, Zhang LL, Sun HJ, Liu TY, Zhu GQ. Melanocortin 3/4 receptors in paraventricular nucleus modulate sympathetic outflow and blood pressure. Exp Physiol. 2013;98(2):435–43.
Haynes WG, Morgan DA, Djalali A, Sivitz WI, Mark AL. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension. 1999;33(1 Pt 2):542–7.
Kuo JJ, Silva AA, Hall JE. Hypothalamic melanocortin receptors and chronic regulation of arterial pressure and renal function. Hypertension. 2003;41(3 Pt 2):768–74.
Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44–52.
da Silva AA, Kuo JJ, Hall JE. Role of hypothalamic melanocortin 3/4-receptors in mediating chronic cardiovascular, renal, and metabolic actions of leptin. Hypertension. 2004;43(6):1312–7.
Tallam LS, Stec DE, Willis MA, da Silva AA, Hall JE. Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension. 2005;46(2):326–32.
Greenfield JR. Melanocortin signalling and the regulation of blood pressure in human obesity. J Neuroendocrinol. 2011;23(2):186–93.
Tallam LS, da Silva AA, Hall JE. Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension. 2006;48(1):58–64.
do Carmo JM, da Silva AA, Wang Z, Fang T, Aberdein N, de Lara Rodriguez CE, et al. Obesity-induced hypertension: brain signaling pathways. Curr Hypertens Rep. 2016;18(7):58.
da Silva AA, do Carmo JM, Wang Z, Hall JE. The brain melanocortin system, sympathetic control, and obesity hypertension. Physiology (Bethesda). 2014;29(3):196–202.
da Silva AA, do Carmo JM, Dubinion JH, Bassi M, Mokhtarpouriani K, Hamza SM, et al. Chronic central nervous system MC3/4R blockade attenuates hypertension induced by nitric oxide synthase inhibition but not by angiotensin II infusion. Hypertension. 2015;65(1):171–7.
do Carmo JM, Bassi M, da Silva AA, Hall JE. Systemic but not central nervous system nitric oxide synthase inhibition exacerbates the hypertensive effects of chronic melanocortin-3/4 receptor activation. Hypertension. 2011;57(3):428–34.
da Silva AA, do Carmo JM, Kanyicska B, Dubinion J, Brandon E, Hall JE. Endogenous melanocortin system activity contributes to the elevated arterial pressure in spontaneously hypertensive rats. Hypertension. 2008;51(4):884–90.
Spradley FT, Palei AC, Anderson CD, Granger JP. Melanocortin-4 receptor deficiency attenuates placental ischemia-induced hypertension in pregnant rats. Hypertension. 2019;73(1):162–70.
Samuelsson AS, Mullier A, Maicas N, Oosterhuis NR, Eun Bae S, Novoselova TV, et al. Central role for melanocortin-4 receptors in offspring hypertension arising from maternal obesity. Proc Natl Acad Sci U S A. 2016;113(43):12298–303.
• do Carmo JM, da Silva AA, Moak SP, da Silva FS, Spradley FT, Hall JE. Role of melanocortin 4 receptor in hypertension induced by chronic intermittent hypoxia. Acta Physiol (Oxf). 2019;225(4):e13222. This study demonstrated that MC4R does not play an important role in hypertension induced by chronic intermittent hypoxia. This is a surprising finding since most, if not all, previous studies that examined MC4R’s role in BP regulation in animal models known to be associated with increased SNS activity showed significant impact of MC4R antagonism to reduce SNS activity and lower BP.
Takahashi K, Ueda S, Kobayashi T, Nishiyama A, Fujisawa Y, Sugaya T, et al. Chronic intermittent hypoxia-mediated renal sympathetic nerve activation in hypertension and cardiovascular disease. Sci Rep. 2018;8(1):17926.
Schiavon CA, Ikeoka D, Santucci EV, Santos RN, Damiani LP, Bueno PT, et al. Effects of bariatric surgery versus medical therapy on the 24-hour ambulatory blood pressure and the prevalence of resistant hypertension. Hypertension. 2019;73(3):571–7.
Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 2011;13(2):195–204.
Tao YX. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31(4):506–43.
Ward KR, Bardgett JF, Wolfgang L, Stocker SD. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension. 2011;57(3):435–41.
Sohn JW, Harris LE, Berglund ED, Liu T, Vong L, Lowell BB, et al. Melanocortin 4 receptors reciprocally regulate sympathetic and parasympathetic preganglionic neurons. Cell. 2013;152(3):612–9.
Berglund ED, Liu T, Kong X, Sohn JW, Vong L, Deng Z, et al. Melanocortin 4 receptors in autonomic neurons regulate thermogenesis and glycemia. Nat Neurosci. 2014;17(7):911–3.
da Silva AA, do Carmo JM, Freeman JN, Tallam LS, Hall JE. A functional melanocortin system may be required for chronic CNS-mediated antidiabetic and cardiovascular actions of leptin. Diabetes. 2009;58(8):1749–56.
Kay EI, Botha R, Montgomery JM, Mountjoy KG. hMRAPalpha, but not hMRAP2, enhances hMC4R constitutive activity in HEK293 cells and this is not dependent on hMRAPalpha induced changes in hMC4R complex N-linked glycosylation. PLoS One. 2015;10(10):e0140320.
Newman EA, Chai BX, Zhang W, Li JY, Ammori JB, Mulholland MW. Activation of the melanocortin-4 receptor mobilizes intracellular free calcium in immortalized hypothalamic neurons. J Surg Res. 2006;132(2):201–7.
• Li YQ, Shrestha Y, Pandey M, Chen M, Kablan A, Gavrilova O, et al. G(q/11)alpha and G(s)alpha mediate distinct physiological responses to central melanocortins. J Clin Invest. 2016;126(1):40–9. This study demonstrates that MC4R can activate different types of G-coupled proteins to differentially regulate appetite, linear growth, thermogenesis, and blood pressure. More specifically, it was shown that deletion of G(q/11α) protein in PVN neurons makes the mice unresponsive to the appetite suppressant actions of MC4R agonists delivered into the PVN, while (s)α deletion in the same group of neurons abolishes MC4R agonistic-induced elevations in blood pressure.
Litt MJ, Cone RD, Ghamari-Langroudi M. Characterization of MC4R regulation of the Kir7.1 channel using the Tl(+) flux assay. Methods Mol Biol. 2018;1684:211–22.
• Anderson EJP, Ghamari-Langroudi M, Cakir I, Litt MJ, Chen V, Reggiardo RE, et al. Late onset obesity in mice with targeted deletion of potassium inward rectifier Kir7.1 from cells expressing the melanocortin-4 receptor. J Neuroendocrinol. 2019;31(1):e12670. This study shows the important contribution of a novel mechanism by which MC4R regulates neuronal firing through activation of Kir7.1 potassium channels. These channels may also contribute to MC4R’s differential regulation of its physiological effects.
Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, et al. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015;100(4):1639–45.
•• Kuhnen P, Clement K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med. 2016;375(3):240–6. This study shows a significant weight loss and improved metabolic profile in obese individuals with POMC deficiency treated with the MC4R agonist, setmelanotide, with minimal impact on blood pressure.
•• Clement K, Biebermann H, Farooqi IS, Van der Ploeg L, Wolters B, Poitou C, et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat Med. 2018;24(5):551–5. The authors describe additional weight loss effects of long-term Setmelanotide treatment in severely obese individuals with POMC deficiency which was associated with improved blood pressure levels.
Liu D, Zhang HG, Zhao ZA, Chang MT, Li Y, Yu J, et al. Melanocortin MC4 receptor agonists alleviate brain damage in abdominal compartment syndrome in the rat. Neuropeptides. 2015;49:55–61.
Gava FN, da Silva AA, Hall JE, do Carmo JM. Chronic central melanocortin 4 receptor activation attenuates cardiac dysfunction after myocardial infarction in rats. Hypertension. 2018;72:A008 (Abstract).
Litt MJ, Okoye GD, Lark D, Cakir I, Moore C, Barber MC, et al. Loss of the melanocortin-4 receptor in mice causes dilated cardiomyopathy. Elife. 2017;6.
Funding
The authors’ research was supported by the National Heart, Lung, and Blood Institute (P01 HL51971), National Institute of General Medical Sciences (P20 GM104357 and U54 GM115428), and National Institute of Diabetes and Digestive and Kidney Diseases (K99DK113280).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human subjects or animals performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Secondary Hypertension: Nervous System Mechanisms
Rights and permissions
About this article

Cite this article
da Silva, A.A., do Carmo, J.M., Wang, Z. et al. Melanocortin-4 Receptors and Sympathetic Nervous System Activation in Hypertension. Curr Hypertens Rep 21, 46 (2019). https://doi.org/10.1007/s11906-019-0951-x
Published:
DOI: https://doi.org/10.1007/s11906-019-0951-x