
Abstract
The activation of the sympathetic nervous system is a major mechanism underlying both human and experimental models of obesity-related hypertension. While insulin and the adipokine leptin have long been thought to contribute to obesity-related neurogenic mechanisms, the evidence is now very strong that they play a major role, shown particularly in animal studies using selective receptor antagonists. There is not just maintenance of leptin’s sympatho-excitatory actions as previously suggested but considerable amplification particularly in renal sympathetic nervous activity. Importantly, these changes are not dependent on short-term elevation or reduction in plasma leptin or insulin, but require some weeks to develop indicating a slow “neural adaptivity” within hypothalamic signalling. These effects can be carried across generations even when offspring are raised on a normal diet. A better understanding of the underlying mechanism should be a high research priority given the prevalence of obesity not just in the current population but also for future generations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
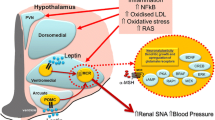
The increase in the prevalence of obesity and the sequelae of hypertension, dyslipidaemia, insulin resistance, diabetes, and cardiovascular disease, affecting both developed and developing countries, has become a major worldwide burden of disease [1]. Analysis of the Framingham study suggests that over two thirds of newly diagnosed cases of hypertension are related to being overweight or obese [2]. The importance of determining the underlying mechanisms of obesity-induced hypertension has, therefore, grown significantly in recent years. This is evidenced by the threefold increase in research articles on obesity hypertension in PubMed over the last ten years while areas such as renovascular or essential hypertension have remained static. While there are many factors contributing to the increase in blood pressure in obese and overweight patients, increasingly, there is recognition of the important contribution of the sympathetic nervous system (SNS) [3–8]. The hypothalamus, well recognised for its role in regulating fluid balance, blood volume, and sodium homeostasis is also the major site for the confluence of neural and hormonal signalling. Perhaps the most relevant of the peripheral signalling molecules are the adipokines leptin and adiponectin, the hormone insulin, and gut hormones such as ghrelin. All of these hormones appear to relay metabolic information to the central nervous system (CNS) via specific receptors particularly within the arcuate nucleus of the hypothalamus. Indeed, there is now a wealth of knowledge about the hypothalamus with respect to the afferent and efferent neural connections, the nature of the neurons within the subregions and the influence of a multitude of blood-borne signalling molecules. The hypothalamus is also a key region for regulating the hormonal and neural response to acute emotional stress. Recent evidence suggests that there may well be overlap between the circuits processing the autonomic responses to stress with those responding to metabolic signalling. Thus, uncovering the cause of CNS dysfunction in obesity is imperative in order to understand the development of obesity-related hypertension. The purpose of this review is to document the progress that has been made in recent years and point the way for future research.
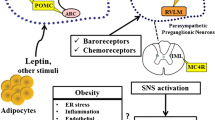
Contribution of the SNS to Obesity Related Hypertension
The landmark early studies from Lansberg and colleagues documented the very strong influence of eating on the activity of the SNS [9]. In a major hypothesis paper, Bray proposed a model that suggested that obesity resulted in reduced SNS activity although the focus of this work involved the metabolic effects of the SNS on the liver, brown adipose tissue, pancreas, and adrenal glands [10]. Changes in SNS activity to different regional beds are not uniform such that a change in activity to one region will not necessarily reflect another [11]. Indeed, obesity is associated with elevations in renal and muscle norepinephrine spillover while cardiac is reduced [12]. Direct measurement of sympathetic activity in humans is technically limited to the muscle bed where there has been a consistent view that activity is elevated in both obese normotensive and hypertensive subjects and particularly in obese hypertensive subjects [13–17]. Interestingly the sympatho-excitation is associated with long- term obesity and the accumulation of visceral fat rather than subcutaneous fat [15, 18, 19]. Analysis of single unit recordings from peroneal nerves by Lambert and colleagues revealed a pattern of sympathetic activation in obese patients that was characterized by greater activity being related to recruited units firing at normal rates [14]. The recruitment of new active units contrasts hypertension in lean patients, which is characterized by an increased probability of firing and increased incidence of multiple firing from active units [14]. While associative studies are simply that, a causal relationship is more difficult to establish, particularly in human obesity. A 10 % weight loss observed seven months after laparoscopic adjustable gastric band surgery resulted in a reduction in blood pressure and also muscle sympathetic activity [20]. Dietary restriction has been shown to reduce total noradrenaline spillover particularly in those with very elevated insulin levels [21]. Perhaps the most convincing evidence comes from studies using ganglionic blockade which show in humans [22], rat [23], rabbit [24], and mouse [25, 26] models that a greater fall in blood pressure was observed in the obese groups compared to the lean controls that essentially abolished the obesity related hypertension (Fig. 1).

A: Hypotensive responses to ganglionic blockade in obese versus lean controls from humans [22], rat [23], rabbit [24], and mouse [25, 26] models. * indicates P < 0.05 between lean and obese. B: Hypotensive responses to administration of insulin antagonist and leptin antagonist after one and three weeks of a normal or high-fat diet in rabbits [27••]. * indicates P < 0.05 between normal diet and high-fat diet
Role of Leptin and Insulin in Obesity-Related Hypertension
A product of the ob gene, leptin is primarily synthesised and secreted from white adipose tissue (WAT) [28] with plasma concentrations of the peptide remaining proportional to adiposity [3]. Leptin has been implicated in numerous physiological functions including wound healing, reproduction, immune response, and glucose homeostasis [29, 30], but its main function is recognised as regulating appetite and metabolism [5]. Leptin increases sympathetic nerve activity (SNA) to brown adipose tissue to increase temperature and expend energy [31]. However, the surprising discovery at the time was that systemic leptin infusions also increased SNA to the kidney (RSNA), adrenal ,and hindlimb [31]. Furthermore, central leptin infusions increase blood pressure [32], RSNA as well as SNS activity to the hindlimb, adrenal, liver, and splanchnic circulation [33–40]. While elevated circulating levels of leptin as observed in obese humans and animals leads to less leptin signalling, and; therefore, reduced satiety, this does not apply to the sympatho-excitatory actions [5, 41]. Halaas and colleagues noted that obese animals were resistant to the appetite suppressant effects of leptin, but they assumed this was due to impaired leptin uptake into the CNS [42]. Rahmouni and colleagues reported that obese agouti mice are resistant to the appetite suppressant actions of leptin but have normal leptin-induced increase in RSNA compared to littermates [43]. Thus, the concept of selective leptin resistance, first introduced by Mark and colleagues [44], outlines the loss of specific functions of leptin in obesity and has been suggested as the cause of obesity-induced hypertension [5, 41]. While there is general consensus for a major role of leptin in diet-induced obesity in experimental animals, there is a contrary view. Tumer and colleagues administered a leptin antagonist by intracerebroventricular (ICV) infusion for two weeks to rats on a high fat diet and did not observe any effect on blood pressure [45]. Furthermore, in human obesity related hypertension, the evidence is mostly associative and, therefore, less convincing [5]. Alvarez and colleagues found that SNA correlated with visceral fat and not subcutaneous fat and not to leptin levels. They suggested that these data do not support leptin causing SNS activation, at least in humans [18]. However, they only studied lean and subcutaneous obese young males, with leptin levels in the latter being quite low and in the range that other studies have observed for young lean subjects [46]. Exciting new findings indicate that individuals with a particular melanocortin receptor type 3 missense variant exhibit a downregulation of receptor expression and show a correlation between plasma leptin and blood pressure [47]. Furthermore, subjects with a loss of function in the melanocortin receptor type 4 receptor have lower blood pressure than matched normal subjects [48]. Mice lacking melanocortin receptor type 4 are obese have normal blood pressure and are not sensitive to salt [49]. A range of studies in different species have shown that central injections of melanocortin type 3 and 4 antagonists reduce blood pressure and sympathetic activity. Dubinion et al., observed a decrease in blood pressure after chronic central infusion of a melanocortin type 3 and 4 antagonist in obese rats [50]. The melanocortin type 4 antagonist JKC-363 given intracerebroventricularly to conscious rabbits abolishes the renal sympatho-excitatory effects of central α-MSH [51]. However, in other studies Li et al., found that both type 3 and 4 receptors antagonists were sympatho-excitatory when applied within the PVN of anesthetised rats, and that antagonists of both receptors were necessary to block the effects of a type 3 and 4 agonist [52]. Using the Zucker rat model of obesity, Do Carmo et al., found that SHU-9119, which is a melanocortin type 3 and 4 antagonist, reduced blood pressure when given chronically ICV [53]. As the leptin receptors are located on arcuate melanocortin neurons projecting into other hypothalamic regions such as the paraventricular and ventrolateral hypothalamus, these findings support the view that signalling in the hypothalamus downstream from leptin’s actions is important in human obesity-related hypertension. Importantly, this is mainly mediated by the melanocortin signalling from arcuate POMC neurons.
Hyperinsulinemia is inextricably associated with obesity due to the presence of insulin resistance and may also contribute to the elevated blood pressure and increased sympathetic activity [54]. Numerous studies have reported the sympatho-excitatory effects of central administration of insulin in experimental models [55–58]. Muntzel and colleagues showed that central administration of insulin to anaesthetised rats activated lumbar but not renal or adrenal SNA [56]. A more recent study found that central insulin administration increased SNA to renal, hindlimb, and brown adipose tissue in normal mice but in agouti obese mice insulin dose dependently increased lumbar SNA whilst renal and brown adipose tissue SNA responses were blunted [59]. Interestingly, a three-day pretreatment with ICV insulin attenuated the sympatho-excitatory effects of leptin in anaesthetised rats [60]. The difficulty with studies in rats and mice comparing SNA between groups is that all measurements are standardised to baseline as 100 %. Thus, an attenuated response or exaggerated response may simply be due to an alteration in basal SNS activity. Furthermore, none of the studies mentioned above determined the contribution of either leptin or insulin to obesity-induced hypertension. The situation in human obesity-related hypertension is less clear than in experimental studies. The Tecumseh offspring study noted that in young subjects with sympathetic overactivity there was a strong correlation between insulin levels and left ventricular hypertrophy [61]. The latter may well reflect long-term levels of blood pressure. Straznicky and colleagues have recently shown that dietary weight loss reduced indices of norepinephrine turnover, and that this was most profound in those with hyperinsulinaemia [21]. Furthermore, a similar weight loss achieved by lap band surgery also reduced SNS activity, insulin, and ambulatory systolic blood pressure but not diastolic blood pressure. Thus, while there are many studies consistent with a putative contribution of hyperinsulinaemia contributing to obesity related hypertension, there are no definitive studies.
The High-Fat Fed Rabbit Model of Obesity
Many studies including ours have shown that rabbits exhibit a typical phenotype of human obesity when fed with a high caloric or a high-fat diet. For instance, when fed an ad libitum high-fat diet, they become obese and develop mild hypertension, hyperinsulinemia, tachycardia, and cardiac hypertrophy compared with rabbits fed a standard diet [62]. The particular advantage of the rabbit is the ability to record RSNA for long periods in a conscious animal and being able to calibrate the total nerve activity such that comparisons can be made between distinct groups [63]. We have shown that feeding rabbits with a 13 % high-fat diet leads to an increase in arterial blood pressure, heart rate, and RSNA as well as plasma noradrenaline, leptin, insulin, visceral fat, and total fat compared to rabbits fed a normal diet (4 % fat) [24, 27••, 64••, 65] (Table 1). Importantly we documented a marked facilitation of the sympatho-excitatory effects of central leptin in these rabbits [65]. Indeed, New Zealand white rabbits on a normal diet showed no sympatho-excitatory responses to doses of up to 100 μg of leptin which contrasts with the Japanese white rabbit that appears to be much more sensitive to the pressor and sympatho-excitatory actions of leptin [40]. While the rabbit fat fed model of obesity has not been widely used compared to rodent and dog models of obesity, it bears a remarkable similarity to human obesity hypertension in many regards particularly with respect to the modest degree of hypertension (9–12 %), increased heart rate, plasma noradrenaline, renal sympathetic activity, hyperleptinaemia, hyperinsulinaemia, and activation of the renin angiotensin system (Table 1). It is worth noting that the 13 % level of fat in the diet given to these rabbits is much less than the current Western diet in the USA which is approximately 32 % according to Cordain et al. [66].
Using radiotelemetry not only to measure blood pressure but also to measure RSNA, we found that an increase in blood pressure, heart rate, and RSNA could be detected on the first day of a high-fat diet [24]. Over the course of the three weeks of high-fat diet, body weight, blood glucose, insulin, leptin, blood pressure, heart rate, and RSNA increased [24]. Switching to a normal diet at the end of three weeks returned glucose, insulin, leptin, and heart rate to baseline levels but blood pressure and RSNA remained elevated. The importance of this finding is that circulating levels of leptin or insulin are not directly related to the hypertension per se although in previous studies we have shown correlations greater than 0.8 between leptin and visceral fat, blood pressure, and RSNA [65]. We suggest that the apparent discrepancy is a result of a delay in the offset of the longer term effects of leptin in “rewiring” the central sympatho-excitatory pathways.
The Transgenerational Cardiovascular Impact of Obesity
Concomitant to the increasing prevalence of obesity in the community is the greater number of obese females of reproductive age [71]. Obesity is typically associated with insulin resistance, and in females this condition leads to increased risk for hypertension, central fat accumulation and adverse outcomes in pregnancy [72]. While there is a known greater risk of infant mortality if the mother is obese, the long-term health impact on the offspring is also very important particularly for cardiovascular outcomes [73]. Velkoska and colleagues showed that overnutrition during suckling in rats led to greater body weight in adulthood and hyperleptinaemia compared to normal nutrition [74]. Using rabbits fed a high-fat diet during pregnancy and lactation, we found that the offspring had elevated blood pressure, heart rate, and RSNA compared with offspring from mothers on a normal diet [75••]. We also observed greater sensitivity to ICV leptin and ghrelin in the offspring, which we suspect is the mechanism underlying the adverse cardiovascular profile in adulthood [75••]. The offspring did show a modest elevation in visceral fat at the time [75••] but the elevation of blood pressure, heart rate, and RSNA were similar to adult animals that were being fed a high-fat diet [27••, 65]. We cannot determine how many of the changes were due to developmental programming [76] or due to the earlier exposure to high-fat nutrition from their mothers. Nevertheless, the potential impact of these findings is enormous if they were translated to the human population where more than forty percent of the pregnancies are from obese or overweight mothers [77].
Effect of Selective Leptin and Insulin Antagonists
The key question is whether the elevated levels of leptin and also insulin, while being sympatho-excitatory, contribute to obesity-induced hypertension. We recently examined this question using doses of specific leptin and insulin antagonists that we found would block the effects of exogenous leptin and insulin, respectively [27••]. We used our 3-week protocol of feeding a high-fat diet fed to rabbits with previously implanted ICV catheters but examined the effect of the antagonists at one week and three weeks of the diet. At these times, there was no discernable difference in plasma levels of insulin or leptin [24]. The central leptin antagonist was without effect after one week of diet, but at three weeks the antagonist normalised blood pressure (to normal diet levels) and markedly reduced RSNA (Fig. 1) [27••]. The insulin antagonist produced a small hypotensive response at both times but had no effect on RSNA (Fig. 1) [27••]. This is consistent with the findings of Muntzel and colleagues, who reported that central doses of insulin increased lumbar SNA but not RSNA [56]. However, as mentioned above, our findings do contrast to those of Tumer and colleagues, who did not observe any effect on blood pressure from a chronically infused leptin antagonist to rats on a high-fat diet [45]. They argued that the dose was sufficient as it blocked the effect of a virally induced elevation of leptin but a comparison of the doses between studies revealed that they used approximately one tenth of the dose used in rabbits, allowing for differences in brain size between rabbits and rats. Importantly, the chronic infusion of the leptin antagonist in the rat induced a marked increase in food intake and body weight, which would very much confound the interpretation. By contrast, our study examined the effects of blocking leptin over a 90 minute period with the peak effect at 30 minutes. We conclude that at least early on in the development of obesity in the rabbit high-fat diet model, there is a major contribution of leptin to the hypertension with a lesser contribution from insulin.
A new Concept of Delayed and Persistent Central Sympatho-Excitation
The current view is that obesity hypertension is due to selective leptin resistance whereby the sympatho-excitatory actions of leptin are preserved while the anorectic actions develop resistance. Our studies suggest that there is little sympatho-excitatory action of leptin centrally in rabbits on a normal diet and that there is a markedly amplified sympatho-excitatory response during sustained fat feeding that is maintained even when the animals return to a normal diet for a week [24] or several weeks [75••]. Furthermore, we have recently discovered that this sympatho-excitation also occurs with ICV administration of alpha-melanocortin stimulating hormone, which is downstream from leptin’s action within the arcuate nucleus [78]. The likely site of action may be within other nuclei of the hypothalamus such as the dorsomedial or ventromedial nuclei [78]. The key features of the concept of central sympatho-excitation are that:
-
1.
The onset is delayed and requires several weeks of a high-fat diet or hyperleptineaemia.
-
2.
The effect is persistent for several weeks after returning to a normal diet.
-
3.
The mechanism involves marked amplification of the central response to leptin.
-
4.
The location may be downstream from leptin signalling at the level of melanocortin pathways.
-
5.
The effect can be transmitted across generations.
These five characteristics, if they were also to be translatable to the human condition, make clear the reason why the obesity epidemic has such a profound impact on the burden of disease in the community. Determining the mechanism and developing counter strategies is therefore a major priority for health researchers.
Contribution of Aversive Stress to Obesity-Related Hypertension
In 2000, a cross-sectional study suggested that stress-induced cortisol secretion contributes to central fat accumulation demonstrating a link between stress and obesity [79]. The following year Bjorntorp proposed a similar hypothesis suggesting a direct link between stress and obesity [80]. He suggested a commonality of central autonomic systems such that repeated activation of the HPA axis and the SNS may cause abdominal obesity and comorbidities. He proposed that chronic stress leading to elevated levels of cortisol as well as low growth and sex steroids promotes the accumulation of visceral fat. Lambert and colleagues showed that single-unit sympathetic nerve firing pattern was correlated to anxiety and depressive states rather than many aspects of metabolic disorder [81]. Also, the pattern of sympathetic activation in hypertensive individuals resembles the defence reaction [82]. Pasquali and colleagues reported greater tachycardia, pressor responses, and ACTH release during a stress test in women with increased visceral adiposity compared to controls [83]. Kunioshi and colleagues found similar increases in muscle SNA to mental stress in obese and lean subjects [84]. These data are in contrast with the finding that subjects with increased BMI and waist to hip ratio had smaller heart rate reactivity to stress [85]. Furthermore, augmented heart rate reactivity to stress was associated with a decreased probability of becoming obese in the ensuing five years [85].
While the cause and effect relationship between stress and obesity has been difficult to sort out, there is a clear overlap between the neural pathways involved in the modulation of stress responses and energy homeostasis [86]. The paraventricular, dorsomedial, and ventromedial hypothalamic regions are major integrative sites for appetite signalling [87] and the sympatho-excitatory actions of leptin [88] as well as being critical for the hormonal and autonomic responses to stress [89, 90]. In the high-fat diet fed rabbit we recently observed a gradual diminution of the sympatho-excitatory response to airjet stress over several weeks and a change in the pattern of the RSNA baroreflex that closely resembled acute stress [24]. We proposed that the elevation of blood pressure and heart rate observed with a high-fat diet is a result of activation of endogenous stress pathways. Therefore, one might only observe a partial further increase during acute stress. While our studies do not shed light on the Bjorntorp hypothesis mentioned earlier, as our rabbits do not have a dietary choice, they confirm the general view of a close relationship between the psychosocial factors, eating and social behaviours and causes of hypertension.
Conclusions
We conclude that there is overwhelming evidence that the cause of obesity-related hypertension is primarily through neurogenic mechanisms. Elevated levels of leptin, insulin, and possibly other adipokines including ghrelin may reprogram the central signalling to produce a state of sympatho-excitation. The evidence to date suggests a major involvement of the melanocortin system. The time frame of the changes depends on the signal. The contribution of insulin to elevating blood pressure in obesity occurs very soon after beginning a high-fat diet while leptin’s contribution takes several weeks to manifest. This difference offers a unique opportunity to explore the contribution of each hormone separately by quickly switching diets in different experimental groups and examining the changes in hypothalamic signalling. We further suggest that the negative aspects of the phenotype such as hypertension can be transmitted across generations, which is alarming considering the prevalence of obesity in pregnancy in some countries. The area which in our view has received little attention is the contribution of chronic aversive stress to the development of obesity and aberrant eating habits. Thus, there is a need to understand more widely the contribution and interaction of “stress” and “metabolic” hormonal signalling in the central nervous system that combine to activate the sympathetic nervous system leading to hypertension in some obese subjects.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Alwan A, World Health Organization. Global Status Report on Noncommunicable Diseases. 2010.
Garrison RJ, Kannel WB, Stokes 3rd J, Castelli WP. Incidence and precursors of hypertension in young adults: the Framingham Offspring Study. Prev Med. 1987;16(2):235–51.
Eikelis N, Esler M. The neurobiology of human obesity. Exp Physiol. 2005;90(5):673–82.
Rahmouni K, Haynes WG, Mark AL. Cardiovascular and sympathetic effects of leptin. Curr Hypertens Rep. 2002;4(2):119–25.
Mark AL. Selective leptin resistance revisited. Am J Physiol Regul Integr Comp Physiol. 2013;305(6):R566–81.
Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285(23):17271–6.
da Silva AA, do Carmo J, Dubinion J, Hall JE. The role of the sympathetic nervous system in obesity-related hypertension. Curr Hypertens Rep. 2009;11(3):206–11.
Hall JE, Zappe DH, AlonsoGalicia M, Granger JP, Brands MW, Kassab SE. Mechanisms of obesity-induced hypertension. News Physiol Sci. 1996;11:255–61.
Landsberg L, Young JB. Fasting, feeding and regulation of the sympathetic nervous system. NEJM. 1978;298(23):1295–301.
Bray GA. Obesity, a disorder of nutrient partitioning: the MONA LISA hypothesis. J Nutr. 1991;121(8):1146–62.
McAllen RM, May CN, Campos RR. The supply of vasomotor drive to individual classes of sympathetic neuron. Clin Exp Hypertens. 1997;19(5–6):607–18.
Vaz M, Jennings G, Turner A, Cox H, Lambert G, Esler M. Regional sympathetic nervous activity and oxygen consumption in obese normotensive human subjects. Circulation. 1997;96(10):3423–9.
Abate NI, Mansour YH, Tuncel M, Arbique D, Chavoshan B, Kizilbash A, et al. Overweight and sympathetic overactivity in black Americans. Hypertension. 2001;38(3):379–83.
Lambert E, Straznicky N, Schlaich M, Esler M, Dawood T, Hotchkin E, et al. Differing pattern of sympathoexcitation in normal-weight and obesity-related hypertension. Hypertension. 2007;50(5):862–8.
Alvarez GE, Beske SD, Ballard TP, Davy KP. Sympathetic neural activation in visceral obesity. Circulation. 2002;106(20):2533–6.
Grassi G, Seravalle G, DellOro R, Turri C, Bolla GB, Mancia G. Adrenergic and reflex abnormalities in obesity-related hypertension. Hypertension. 2000;36(4):538–42.
Lambert E, Straznicky N, Eikelis N, Esler M, Dawood T, Masuo K, et al. Gender differences in sympathetic nervous activity: influence of body mass and blood pressure. J Hypertens. 2007;25(7):1411–9.
Alvarez GE, Ballard TP, Beske SD, Davy KP. Subcutaneous obesity is not associated with sympathetic neural activation. Am J Physiol Heart Circ Physiol. 2004;287(1):H414–8.
Grassi G, Dell'Oro R, Facchini A, Quarti Trevano F, Bolla GB, Mancia G. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J Hypertens. 2004;22(12):2363–9.
Lambert EA, Rice T, Eikelis N, Straznicky NE, Lambert GW, Head GA et al. Sympathetic activity and cardiovascular risk in non-diabetic severely obese patients: The effect of 10 % weight loss. Am J Hypertens. 2014:In press.
Straznicky NE, Lambert EA, Grima MT, Eikelis N, Richards K, Nestel PJ et al. The effects of dietary weight loss on indices of norepinephrine turnover: Modulatory influence of hyperinsulinemia. Obesity (Silver Spring). 2013.
Shibao C, Gamboa A, Diedrich A, Ertl AC, Chen KY, Byrne DW, et al. Autonomic contribution to blood pressure and metabolism in obesity. Hypertension. 2007;49(1):27–33.
Carlson SH, Shelton J, White CR, Wyss JM. Elevated sympathetic activity contributes to hypertension and salt sensitivity in diabetic obese Zucker rats. Hypertension. 2000;35(1 Pt 2):403–8.
Armitage JA, Burke SL, Prior LJ, Barzel B, Eikelis N, Lim K, et al. Rapid onset of renal sympathetic nerve activation in rabbits fed a high-fat diet. Hypertension. 2012;60:163–71.
Rahmouni K, Fath MA, Seo S, Thedens DR, Berry CJ, Weiss R, et al. Leptin resistance contributes to obesity and hypertension in mouse models of Bardet-Biedl syndrome. J Clin Invest. 2008;118(4):1458–67.
Rajapakse NW KF, Fernandez S, Evans RG, Head GA, Kaye DM. L-arginine transporters: a new treatment target in obesity induced hypertension? Hypertension, Abstract from the High Blood Pressure Research Council of Australia. 2013.
Lim K, Burke SL, Head GA. Obesity related hypertension and the role of insulin and leptin in high fat fed rabbits. Hypertension. 2013;61(3):628–34. This landmark report demonstated that elevated blood pressure and renal sympathetic nerve activity induced by a high-fat diet for several weeks are predominantly mediated by delayed central sympathoexcitatory actions of the hormone leptin. Central actions of insulin contribute to a smaller proportion of the hypertension but independently of renal sympathetic activity. Two faculty of 1000 recommendations.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–32.
Frühbeck G. Peripheral actions of leptin and its involvement in disease. Nutr Rev. 2002;60(10 Pt 2):S47–55. discussion S68-84, 5–7.
Haque MS, Minokoshi Y, Hamai M, Iwai M, Horiuchi M, Shimazu T. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes. 1999;48(9):1706–12.
Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI. Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100(2):270–8.
Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension. 1998;31(1 Pt 2):409–14.
Dunbar JC, Hu Y, Lu H. Intracerebroventricular leptin increases lumbar and renal sympathetic nerve activity and blood pressure in normal rats. Diabetes. 1997;46(12):2040–3.
Dunbar JC, Lu H. Leptin-induced increase in sympathetic nervous and cardiovascular tone is mediated by proopiomelanocortin (POMC) products. Brain Res Bull. 1999;50(3):215–21.
Haynes WG, Morgan DA, Djalali A, Sivitz WI, Mark AL. Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension. 1999;33(1 Pt 2):542–7.
Li B, Shi Z, Cassaglia PA, Brooks VL. Leptin acts in the forebrain to differentially influence baroreflex control of lumbar, renal, and splanchnic sympathetic nerve activity and heart rate. Hypertension. 2013;61(4):812–9.
Warne JP, Alemi F, Reed AS, Varonin JM, Chan H, Piper ML, et al. Impairment of central leptin-mediated PI3K signaling manifested as hepatic steatosis independent of hyperphagia and obesity. Cell Metab. 2011;14(6):791–803.
Satoh N, Ogawa Y, Katsuura G, Numata Y, Tsuji T, Hayase M, et al. Sympathetic activation of leptin via the ventromedial hypothalamus: leptin-induced increase in catecholamine secretion. Diabetes. 1999;48(9):1787–93.
Tang-Christensen M, Havel PJ, Jacobs RR, Larsen PJ, Cameron JL. Central administration of leptin inhibits food intake and activates the sympathetic nervous system in rhesus macaques. J Clin Endocrinol Metab. 1999;84(2):711–7.
Matsumura K, Abe I, Tsuchihashi T, Fujishima M. Central effects of leptin on cardiovascular and neurohormonal responses in conscious rabbits. Am J Physiol Regul Integr Comp Physiol. 2000;278(5):R1314–20.
Mark AL, Correia ML, Rahmouni K, Haynes WG. Selective leptin resistance: a new concept in leptin physiology with cardiovascular implications. J Hypertens. 2002;20(7):1245–50.
Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci U S A. 1997;94(16):8878–83.
Rahmouni K, Haynes WG, Morgan DA, Mark AL. Selective resistance to central neural administration of leptin in agouti obese mice. Hypertension. 2002;39(2 Part 2 Suppl S):486–90.
Aizawa-Abe M, Ogawa Y, Masuzaki H, Ebihara K, Satoh N, Iwai H, et al. Pathophysiological role of leptin in obesity-related hypertension. J Clin Invest. 2000;105(9):1243–52.
Tumer N, Erdos B, Matheny M, Cudykier I, Scarpace PJ. Leptin antagonist reverses hypertension caused by leptin overexpression, but fails to normalize obesity-related hypertension. J Hypertens. 2007;25(12):2471–8.
Lambert E, Sari CI, Dawood T, Nguyen J, McGrane M, Eikelis N, et al. Sympathetic nervous system activity is associated with obesity-induced subclinical organ damage in young adults. Hypertension. 2010;56(3):351–8.
Alsmadi O, Melhem M, Hebbar P, Thareja G, John SE, Alkayal F, et al. Leptin in association with common variants of MC3R mediates hypertension. Am J Hypertens. 2014;27(7):973–81.
Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44–52.
Tallam LS, Stec DE, Willis MA, da Silva AA, Hall JE. Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension. 2005;46(2):326–32.
Dubinion JH, da Silva AA, Hall JE. Enhanced blood pressure and appetite responses to chronic central melanocortin-3/4 receptor blockade in dietary-induced obesity. J Hypertens. 2010;28(7):1466–70.
Matsumura K, Tsuchihashi T, Abe I, Iida M. Central alpha-melanocyte-stimulating hormone acts at melanocortin-4 receptor to activate sympathetic nervous system in conscious rabbits. Brain Res. 2002;948(1–2):145–8.
Li P, Cui BP, Zhang LL, Sun HJ, Liu TY, Zhu GQ. Melanocortin 3/4 receptors in paraventricular nucleus modulate sympathetic outflow and blood pressure. Exp Physiol. 2013;98(2):435–43.
do Carmo JM, da Silva AA, Rushing JS, Hall JE. Activation of the central melanocortin system contributes to the increased arterial pressure in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2012;302(5):R561–7.
Landsberg L. Obesity, metabolism, and hypertension. Yale J Biol Med. 1989;62(5):511–9.
Holt SJ, York DA. Interaction of intracerebroventricular insulin and glucose in the regulation of the activity of sympathetic efferent nerves to brown adipose tissue in lean and obese Zucker rats. Brain Res. 1989;500(1–2):384–8.
Muntzel M, Morgan D, Mark A, Johnson A. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. Am J Physiol Regul Integr Comp Physiol. 1994;36:R1350–R35.
Cassaglia PA, Hermes SM, Aicher SA, Brooks VL. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol. 2011;589(Pt 7):1643–62.
Pricher MP, Freeman KL, Brooks VL. Insulin in the brain increases gain of baroreflex control of heart rate and lumbar sympathetic nerve activity. Hypertension. 2008;51(2):514–20.
Morgan DA, Rahmouni K. Differential effects of insulin on sympathetic nerve activity in agouti obese mice. J Hypertens. 2010;28(9):1913–9.
Dunbar JC, Lu H. Chronic intracerebroventricular insulin attenuates the leptin-mediated but not alpha melanocyte stimulating hormone increase in sympathetic and cardiovascular responses. Brain Res Bull. 2000;52(2):123–6.
Palatini P, Majahalme S, Amerena J, Nesbitt S, Vriz O, Michieletto M, et al. Determinants of left ventricular structure and mass in young subjects with sympathetic over-activity. The Tecumseh Offspring Study. J Hypertens. 2000;18(6):769–75.
Carroll JF, Dwyer TM, Grady AW, Reinhart GA, Montani JP, Cockrell K, et al. Hypertension, cardiac hypertrophy, and neurohumoral activity in a new animal model of obesity. Am J Physiol Heart Circ Physiol. 1996;271(1 Pt 2):H373–8.
Burke SL, Head GA. Method for in vivo calibration of renal sympathetic nerve activity in rabbits. J Neurosci Methods. 2003;127:63–74.
Burke SL, Prior LJ, Lukoshkova EV, Lim K, Barzel B, Davern P, et al. Reduced preprandial dipping accounts for rapid elevation of blood pressure and renal sympathetic nerve activity in rabbits fed a high-fat diet. Chronobiol Int. 2013;30(5):726–38. This study identified that elevated blood pressure induced by a high-fat diet from the first day is mediated by increased sympathetic nerve activity due to a reduction in preprandial dipping and that increased calories, glucose, insulin, and leptin may account for early changes whilst lon- term loss of dipping may be related to increased sensitivity of sympathetic pathways.
Prior LJ, Eikelis N, Armitage JA, Davern PJ, Burke SL, Montani J-P, et al. Exposure to a high-fat diet alters leptin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension. 2010;55(4):862–8.
Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. 2005;81(2):341–54.
Jones DW. Body weight and blood pressure. Effects of weight reduction on hypertension. Am J Hypertens. 1996;9(8):50s–4.
Carroll JF, Huang M, Hester RL, Cockrell K, Mizelle HL. Hemodynamic alterations in hypertensive obese rabbits. Hypertension. 1995;26(3):465–70.
Carroll JF, Summers RL, Dzielak DJ, Cockrell K, Montani JP, Mizelle HL. Diastolic compliance is reduced in obese rabbits. Hypertension. 1999;33(3):811–5.
Antic V, Kiener-Belforti F, Tempini A, Montani J-P. Obesity-induced hypertension in rabbits is not salt-sensitive. J Hypertens. 1999;17 Suppl 3:S209.
Taylor PD, Samuelsson AM, Poston L. Maternal obesity and the developmental programming of hypertension: a role for leptin. Acta Physiol (Oxf). 2014;210(3):508–23.
Catalano PM. Obesity, insulin resistance, and pregnancy outcome. Reproduction. 2010;140(3):365–71.
Meehan S, Beck CR, Mair-Jenkins J, Leonardi-Bee J, Puleston R. Maternal obesity and infant mortality: a meta-analysis. Pediatrics. 2014
Velkoska E, Cole TJ, Morris MJ. Early dietary intervention: long-term effects on blood pressure, brain neuropeptide Y, and adiposity markers. Am J Physiol Endocrinol Metab. 2005;288(6):E1236–43.
Prior L, Davern P, Burke S, Lim K, Armitage J, Head G. Exposure to a high fat diet during development alters leptin and ghrelin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension. 2014;63(2):338–45. This landmark study revealed that offspring from mothers consuming a high-fat diet exhibit an adverse cardiovascular profile in adulthood including hypertension and increased renal sympathetic activity due to altered central hypothalamic sensitivity to leptin and ghrelin. Faculty of 1000 recommendation.
Henry SL, Barzel B, Burke SL, Head GA, Armitage JA. Developmental origins of obesity related hypertension. Clin Exp Pharmacol Physiol. 2012;39:799–806.
Athukorala C, Rumbold AR, Willson KJ, Crowther CA. The risk of adverse pregnancy outcomes in women who are overweight or obese. BMC Pregnancy Childbirth. 2010;10:56.
Barzel B, Burke S, Armitage J, Head G, editors. Alpha melanocortin stimulating hormone actions at the ventromedial hypothalamus increase renal sympathetic actvity in fat fed rabbits. Experimental Biology; 2013 April 2013; Boston, USA.
Epel ES, McEwen B, Seeman T, Matthews K, Castellazzo G, Brownell KD, et al. Stress and body shape: stress-induced cortisol secretion is consistently greater among women with central fat. Psychosom Med. 2000;62(5):623–32.
Bjorntorp P. Do stress reactions cause abdominal obesity and comorbidities? Obes Rev. 2001;2(2):73–86.
Lambert E, Dawood T, Straznicky N, Sari C, Schlaich M, Esler M, et al. Association between the sympathetic firing pattern and anxiety level in patients with the metabolic syndrome and elevated blood pressure. J Hypertens. 2010;28(3):543–50.
Rumantir MS, Vaz M, Jennings GL, Collier G, Kaye DM, Seals DR, et al. Neural mechanisms in human obesity-related hypertension. J Hypertens. 1999;17(8):1125–33.
Pasquali R, Anconetani B, Chattat R, Biscotti M, Spinucci G, Casimirri F, et al. Hypothalamic-pituitary-adrenal axis activity and its relationship to the autonomic nervous system in women with visceral and subcutaneous obesity: effects of the corticotropin-releasing factor/arginine-vasopressin test and of stress. Metab Clin Exp. 1996;45(3):351–6.
Kuniyoshi FHS, Trombetta IC, Batalha LT, Rondon M, Laterza MC, Gowdak MMG, et al. Abnormal neurovascular control during sympathoexcitation in obesity. Obes Res. 2003;11(11):1411–9.
Carroll D, Phillips AC, Der G. Body mass index, abdominal adiposity, obesity, and cardiovascular reactions to psychological stress in a large community sample. Psychosom Med. 2008;70(6):653–60.
Korner P. Essential hypertension and its causes: neural and non neural mechanisms. New York: Oxford University Press; 2007.
Elmquist JK. Hypothalamic pathways underlying the endocrine, autonomic, and behavioral effects of leptin. Physiol Behav. 2001;74(4–5):703–8.
Marsh AJ, Fontes MA, Killinger S, Pawlak DB, Polson JW, Dampney RA. Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension. 2003;42(4):488–93.
McDougall SJ, Widdop RE, Lawrence AJ. Central autonomic integration of psychological stressors: focus on cardiovascular modulation. Auton Neurosci. 2005;123(1–2):1–11.
De Matteo R, Head GA, Mayorov DN. Angiotensin II in dorsomedial hypothalamus modulates cardiovascular arousal caused by stress but not feeding in rabbits. Am J Physiol Regul Integr Comp Physiol. 2006;290(1):R257–64.
Acknowledgments
This work was supported by grants from the National Health & Medical Research Council of Australia (NHMRC; APP526618 and APP1043205). The study was supported, in part, by the Victorian Government’s Operational Infrastructure Support Program. G.A. Head was funded by an NHMRC Fellowship (APP1002186). K. Lim was funded by an NHMRC Postdoctoral Fellowship (APP1053928). P.J. Davern was funded an NHMRC/National Heart Foundation Postdoctoral Fellowship (APP1012881).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Geoffrey A. Head, Kyungjoon Lim, Benjamin Barzel, Sandra L. Burke, and Pamela J. Davern have received a grant from the National Health & Medical Research Council of Australia.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Hypertension and the Brain
Rights and permissions
About this article

Cite this article
Head, G.A., Lim, K., Barzel, B. et al. Central Nervous System Dysfunction in Obesity-Induced Hypertension. Curr Hypertens Rep 16, 466 (2014). https://doi.org/10.1007/s11906-014-0466-4
Published:
DOI: https://doi.org/10.1007/s11906-014-0466-4