
Abstract
The pulmonary circulation is a high-flow and low-pressure circuit. The functional state of the pulmonary circulation is defined by pulmonary vascular pressure–flow relationships conforming to distensible vessel models with a correction for hematocrit. The product of pulmonary arterial compliance and resistance is constant, but with a slight decrease as a result of increased pulsatile hydraulic load in the presence of increased venous pressure or proximal pulmonary arterial obstruction. An increase in left atrial pressure is transmitted upstream with a ratio ≥1 for mean pulmonary artery pressure and ≤1 the diastolic pulmonary pressure. Therefore, the diastolic pressure gradient is more appropriate than the transpulmonary pressure gradient to identify pulmonary vascular disease in left heart conditions. Exercise is associated with a decrease in pulmonary vascular resistance and an increase in pulmonary arterial compliance. Right ventricular function is coupled to the pulmonary circulation with an optimal ratio of end-systolic to arterial elastances of 1.5–2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pulmonary circulation, as a separate high-flow, low-pressure system, is the end result of an evolutionary process aimed at the optimization of gas exchange of endothermic birds and mammals [1••]. Evolution from ancestors of fishes to amphibians, reptiles and finally birds and mammals has led to progressively greater oxygen consumption requiring a thinner pulmonary blood–gas barrier. Preservation of the integrity of this barrier has been made possible by the complete separation of the pulmonary from the systemic circulation. This evoluton has been accompanied by a progressive unloading and reshaping of the right ventricle (RV) as a thin-walled flow generator unable to adapt to brisk increases in afterload.
Pulmonary Vascular Pressures and Resistance
Pulmonary Artery Pressures and Blood Flow
The pulmonary circulation is characterized by an inflow pressure, or pulmonary artery pressure (Ppa), an outflow pressure, or left atrial pressure (Pla) and a pulmonary blood flow (Q) approximately equal to systemic cardiac output. Pulmonary vascular pressures and flows are pulsatile. However, a simple and clinically useful description of the functional state of the pulmonary circulation may be provided by a calculation of pulmonary vascular resistance (PVR) from mean values of Ppa (mPpa), Pla and Q.
Measurements of pulmonary vascular pressures and cardiac output are usually performed during a catheterization of the right heart with a fluid-filled balloon-tipped thermodilution catheter [2]. This procedure allows for the estimation of Pla from a balloon-occluded (Ppao) or wedged (Ppw) Ppa and Q by thermodilution.
Fluid-filled catheters measure vascular pressures with a zero-levelled external manometer. The best reference is the hydrostatic indifferent point, at the level of the tricuspid valve, where pressure is independent of body position. This is midchest or 5 cm below the sternal angle in the supine position, with mid-axillary intersection at the two inferior fourths of the rib cage to consider when measurements are in the upright position. Zero leveling at the catheterization table is associated with an overestimation of Ppa and Ppw.
Measurements are generally performed at end-expiration, when the lungs are at functional residual capacity. Lung volumes below or above are associated with increased PVR, because of predominant increased in alveolar vessel resistance at high lung volumes, and increased extra-alveolar vessel resistance at lower lung volumes [3]. Thus patients with obstructed airways as a cause of increased functional residual capacity may present with an increased PVR. Furthermore, these patients have important intathoracic pressure swings, which are predominantly positive during expiration [4•]. Measurements at end-expiration may then overestimate Ppa and Ppw.
Sometimes a measurement of Pla or Ppao cannot be obtained, and a total PVR (TPVR) is calculated as
This is may be an acceptable approximation as long as Pla is low, but in all circumstances, TPVR compared to PVR overestimates the extent of pulmonary vascular disease. On the other hand, TPVR is a better estimate of RV afterload than PVR because the RV is exposed to mPpa, not the difference between mPpa and Pla.
Pulmonary Capillary Pressure
Wedged or occluded Ppa measurements (Ppw) are acceptable estimates of Pla, with an average gradient of 3 mmHg between the Ppw and left-ventricular end-diastolic pressure [5]. Micropuncture studies have shown that pulmonary capillary pressure (Ppc) is higher than Ppw, about halfway between arterial and venous pressures [6]. Thus wedged or occluded Ppa should not be called “capillary” or “capillary-wedge” pressure. Estimates of Ppc can be obtained from the analysis of Ppa decay curves after single arterial balloon occlusion [7]. These curves present with an initial fast portion, corresponding to the interruption of flow through the arterial resistance, and a subsequent slow portion corresponding to the emptying of the capillary venous resistance. The intersection of the fast and slow portions identifies Ppc.
The normal longitudinal distribution of resistances is 60 % arterial and 40 % capillary + venous resistance [7]. Ppc can thus be estimated from an equation initially proposed by Gaar [8]:
The site of most pulmonary vascular diseases is at the resistive arterioles. Accordingly, the longitudinal disdtribution of resistances remains unchanged and the Gaar equation generally applicable. An important exception is proximal chronic thromboembolic pulmonary hypertension (CTEPH) [9]. In these patients, an increased steep initial portion of the Ppa decay curve after single occlusion may help to identify predominantly proximal lesions that are amenable to surgical desobstruction [10, 11•].
The Calculation of PVR
A vascular resistance calculation is an extrapolation to a vascular system of a physical law defining laminar flows of Newtonian fluids through non-distensible, straight cylindric tubes, originally proposed by Poiseuille and mathematically formulated by Hagen. The Hagen–Poiseuille law states that resistance R to flow of a single tube is equal to the product of the length l of the tube and viscosity η and a constant 8 divided by the product of π and the fourth power of the internal radius r. More generally, R can be calculated as a pressure drop ΔP to flow Q ratio:
The ratio of pressure drop to flow through an entire vascular bed accounts for the resistances in series and in parallel of the individual vessels. The fact that r in the equation is to the fourth power explains why resistance is exquisitely sensitive to small changes in caliber of these small vessels. For example, a 10 % change in radius results in almost 50 % change in resistance. Accordingly, PVR is a good indicator of the state of constriction or dilatation of pulmonary resistive vessels and is useful for detecting changes in arteriolar vessel caliber due to changes in tone and/or structure.
Effects of Age, Sex and Body Position
The limits of normal of resting pulmonary vascular pressures and flows as derived from invasive measurements in 60 resting supine young adult healthy volunteers [12–15] are shown in Table 1.
In that study population, cardiac output was lower in women, who are smaller than men, and thus PVR in women was higher. However, there were no sex differences in pulmonary hemodynamics after correction for body dimensions. These data have been confirmed by a recent review of invasive measurements reported in a total of 1,187 individuals, of whom 225 were identified as women and 717 as men [16].
Aging is associated with an increase in PVR. This is due to a slight increase in mPpa and a more important decrease in cardiac output leading to a doubling of PVR over a a period of five decades of life [17, 18]. However, measurements in healthy old individuals are few, so that the exact limits of normal of the pulmonary circulation as a function of age are not exactly known [16].
Body position affects PVR through associated changes in systemic venous return. In the upright position, Pla, right atrial pressure (Pra) and cardiac output are lower than in the supine position. Because of pulmonary vascular de-recruitment, mPpa remains essentially the same. Accordingly, PVR is increased. This difference in upright vs. supine PVR is important to keep in mind when examining PVR changes during exercise performed upright as compared to supine [19••].
Effects of Pulmonary Blood Flow
The inherent assumption in a PVR calculation is that the mPpa–flow relationship is constant and crosses the pressure axis at a value equal to Pla. Accordingly, PVR is assumed to be constant, independent of the absolute pressure or flow.
The relationship between (mPpa − Pla) and Q has been shown to be reasonably well described by a linear approximation over a limited “physiological” range of flows, with a zero extrapolated pressure intercept in well oxygenated supine lungs. However, cardiac and respiratory diseases and hypoxia may increase both the slope and the extrapolated intercepts of multipoint mPpa − Q plots. This is explained by a combination of changes in incremental resistance and vascular recruitment in a “waterfall” model made of parallel collapsible vessels with a distribution of closing pressures initially proposed by Permutt [20]. The waterfall analogy refers to the fact that the flow rate (Q) over a waterfall is independent of its height (the pressure difference between upstream and downstream). A typical characteristic of the waterfall model is a functional dissociation between inflow and outflow pressure as long as the latter varies within a range of values that are below a mean “closing pressure”. This is a feature of de-recruited upright upper lung zone I described by West [21], and has been reported in pulmonary hypertension on acute lung injury [22, 23] and in occasional cases of pulmonary hypertension on terminal heart failure, after cardiac transplantation [24].
However the waterfall model ignores the natural distensibility if the pulmonary arterioles, which accounts for the slight curvilinearity seen on multi-point mPpa − Q plots, and wrongly predicts an increased closing pressure from the linear fit of multiple mPpa − Q coordinates in the presence of pulmonary embolism or increased hematocrit [25].
The distensibility of pulmonary resistive vessels has been shown to be of 2 % of diameter change per millimetre of mercury of distending pressure over a wide range of vascular segments and animal species [26]. Linehan integrated resistive vessel distensibility in a simple model allowing for more realistc prediction of pulmonary vascular pressures over a wide range of flows and hematocrits [24]. In this model PVR at a normal hematocrit is calculated as:
The coefficient α is the distensibility factor expressed in % increase in diameter D 0 per millimetre of mercury increase in pressure:
An interesting application of this approach is that the distensibility coefficient α can be recalculated from given sets of Ppa, Pla and Q measurements.
Reeves used reported pulmonary hemodynamic data at rest and during exercise, in healthy volunteers to recalculate α, and found it equal to 2 ± 0.2 %/mmHg in normoxia, a value strikingly similar to that of previous in vitro measurements on isolated vessel segments [27]. Even though the individual data available to him for analysis was limited, he was able to show that α tends to decrease with aging, or with chronic but not acute hypoxic exposure.
Similar values of α have been calculated in a series of noninvasive exercise stress Doppler echocardiographic studies of the normal pulmonary circulation [28–31] and recently confirmed by invasive measurements [32••]. This data has allowed for an improved definition of the limits of normal of the pulmonary circulation at exercise [32••]. Furthermore, α was shown to be lower in men compared to pre-menopausal women [29]. The same noninvasive approach has confirmed Reeves’ observation of a decrease of α with aging [29] and with chronic hypoxic exposure [32••]. There is a suggestion that a decrease in α calculated from noninvasive multiple mPpa − Q coordinates could be sensitive to early pulmonary vascular disease, such as in healthy carriers of the BMPR-2 mutation, which predisposes to pulmonary arterial hypertension (PAH) [33].
The distansibility model of Linehan can also be used to estimate the effects of pulmonary vascular obstruction on mPpa over a wide range of cardiac outputs. The results of this modeling, shown in Fig. 1, agree with previous predictions using an alternative modified viscoelastic model and comparison with angiographic obstruction in a canine model of pulmonary embolism [34]. Thus mPpa reaches 25 mmHg at 50 % obstruction of the pulmonary circulation at a cardiac output of 5 L/min. The current definition of pulmonary hypertension based on a mPpa > 25 mmHg identifies patients with already advanced pulmonary vascular disease [35].

Mean pulmonary artery pressure (Ppa) − cardiac output (Q) relationships predicted by the distensibility model of Linehan at increasing levels of obstruction. An increase in mean Ppa above 25 mmHg corresponding to the definition of pulmonary hypertension occurs after approximately 50 % obstruction of the pulmonary vascular bed. A mean Ppa of 50 mmHg as seen in severe pulmonary hypertension corresponds to 80 % obstruction. These estimates are for a Q of 5 L/min
Viscosity
In the Poiseuille–Hagen equation, resistance is directly proportional to viscosity. The viscosity of the blood is mainly determined by the hematocrit.
The most often used reference equation to estimate the impact of blood viscosity on resistance was reported by Whittaker and Winston based on studies on hindlimb vessels [36]. The equation relates linearly resistance to hematocrit, and allows to re-calculate resistance at a normal reference value of hematocrit of 45 %:
where R 0 is resistance at a hematocrit (HCT) of 45 % and φ the measured hematocrit.
Linehan integrated an exponential relationship in his distensible model of the pulmonary circulation to fit isolated perfused lung measurements at variable levels of hemodilution or concentration [25]:
Both equations allow for similar adjustments of PVR in patients with abnormally high or low hematocrits, and thus provide more realistic estimates of the extent of pulmonary vascular disease [37]. It was recently shown that corrections for hematocrit smoothens ethnic differences in the pulmonary circulation of high-altitude Andean vs. Himalayans populations [38]. In terms of resting PVR, normalizing PVR for a hematocrit may increase PVR by up to 2 Wood units in severely anemic patients, for example with sickle-cell disease.
Left Atrial Pressure and the Transpulmonary Pressure Gradient
An increase in Pla is transmitted upstream to mPpa. The PVR equation assumes that this is in a 1/1 ratio at any given level of Q. A chronic increase in Pla may induce pulmonary vascular remodeling, and therefore lead to an “out of proportion” increase in mPpa [39]. For this reason, clinicians like to reason in terms of a transpulmonary pressure gradient (TPG) for the differential diagnosisd of purely passive increase in mPpa and increased mPpa resulting from pulmonary vascular disease [40]. The TPG is equal to the difference between mPpa and Pla.
The upper limit of normmal of the TPG is usually assumed to be of 12 mmHg [40]. This corresponds to a PVR of 1.5 Wood units at a cardiac output at the upper limit of normal of 8 L/min.
However, it has been recently realized that the TPG is often higher than 12 mmHg in patients with left heart failure in whom purely passive upstream transmission could be demonstrated by observing an acute return of the TPG to <12 mmHg after active diuresis or after a cardiac transplantation [24].
In steady-flow conditions, an increase in Pla is transmitted upstream in a slightly less than 1/1 ratio because the pulmonary resistive vessels are distensible. In pulsatile flow conditions, an increased Pla increases pulse pressure, or difference between systolic and diastolic Ppa. Furthermore, the TPG increases with Q because an increase in flow increases mPpa more than Pla. Thus any increase in Pla or Q above normal increases the TPG. These problems are limited or even avoided by using the gradient between diastolic Ppa and Pla, or the diastolic pressure gradient (DPG) instead [41••]. The upper limit of normal of the DPG in young athletic adults is apprtoximately 5 mmHg [42]. A higher cut-off value may be more reasonable in older patients with left heart failure [43].
The DPG has been proposed to be at the basis of decision tree to identify pulmonary vascular disease in patients with a high Ppw, as illustrated in Fig. 2.

Diastolic pressure–derived decision tree for the diagnosis of pulmonary vascular disease. See text for abbreviations. From Naeije et al. [41••]. Reproduced with permission of the European Respiratory Society. Eur Respir J Jan 2013 41: 217–213; doi:10.1183/09031936.00074312
Exercise
Exercise stresses the pulmonary circulation by an increase in cardiac output. The average slopes of mPpa − Q relationships at exercise will be of 1 mmHg/L/min in young adults and up to 2.5 mmHg/L/min after 5 to 6 decades of life [19••, 32••, 44, 45]. An increase in Pla above the upper limits of normal contributes to the increase in mPpa at high cardiac outputs, in the range of 15–20 L/min/min and more [46]. Exercise-induced cardiac output is associated with a slight decrease in PVR which is entirely explained by resistive vessel distension [19••, 32••]. Exercise mPpa − Q relationships are not affected by body position.
The limits of normal of the pulmonary circulation at exercise are shown in Fig. 3. It can be seen that mPpa does normally exceed 30 mmHg at a cardiac output <10 L/min. Upper limits of normal can also be defined by a slope of mPpa–Q of 3 mmHg/L/min, or a TPVR at maximum exercise of 3 Wood unit [32••].

Pulmonary artery pressure–flow relationships in healthy subjects, with limits of normal indicated by the shaded area. See text for abbreviations
After exercise, mPpa and Q rapidly return to resting values [28]. This decreases the relevance of post-exercise measurements as a reflection of exercise-induced changes. On the other hand, the workload–Q relationship varies considerably from one subject to another [29]. It is therefore preferable to express mPpa at exercise as a function of cardiac output rather than of workload to define the functional state of the pulmonary circulation.
It must be underscored that abnormal exercise pulmonary vascular responses may be due to either increased PVR or Pla. Therefore, the identification of a mPpa–Q relationship >3 mmHg/L/min requires a differential diagnostic work-up [32••].
Pulsatile Flow Pulmonary Hemodynamics
The study of the pulmonary circulation as a steady-flow system is a simplification. Pulmonary pulse pressure is in the order of mPpa, as compared less than half of it in the systemic circulation. Instantaneous pulmonary blood flow varies from a maximum at mid-systole to around zero in diastole.
An analysis of pulsatile flow hemodynamics can be made by a spectral analysis of pressure and flow waves and a calculation of pulmonary vascular impedance (PVZ). PVZ is a ratio of pulsatile Ppa on pulsatile Q. This approach allows for the quantification of PVR, pulmonary arterial elastance (Ca) and wave reflections to pressure and flow wave morphology, and pulmonary arterial hydraulic load as a measure of RV afterload [47, 48].
There has been recently a series of studies which showed that the product of PVR and Ca, or the time constant (RC-time) of the pulmonary circulation remains constant over a wide range of severities, etiologies and treatments of pulmonary hypertension [49–51]. This remarkable property of the pulmonary circulation, which had been previously reported but largely unnoticed [52] has two consequences. The first is that Ca becomes a more important determinant of RV afterload than PVR when mPpa and PVR are only modestly elevated [53]. The second is that RV oscillatory hydraulic load (Wosc) remains a constant fraction of total load (Wtot) irrespective of Ppa [54].
The RC-time may actually slightly decrease in left ventricular failure because the increase in venous pressure causes the pulmonary circulation to be stiffer at any level of PVR [55•]. Another cause of a slight decrease in the RC-time is proximal pulmonary arterial obstruction, either experimentally by pulmonary arterial banding [56] or in patients with purely proximal CTEPH [57•]. A decreased RC-time is associated with an increased Wosc.
The (near)-constancy of the RC-time explains the reported tight correlation between systolic, diastolic and mPpa in normal subjects and in patients with pulmonary hypertension of various etiologies [58]. Accordingly, mPpa can be calculated from sPpa using a simple formula:
This notion is of practical relevance as non-invasive evaluations of the pulmonary circulation in clinical practice often rely on the measurement of a maximum velocity of tricuspid regurgitation (TR) to calculate a sPpa s using the simplified form of the Bernouilli equation and a measurement or estimate of Pra [59]:
Right Ventricular Function
The RV is functionally coupled to the pulmonary circulation [60]. The structural and functional characteristics of the RV allow for the accomodation of large increases in flow, but are not adapted to rapid increases in afterload. However, the basic laws of the heart remain applicable, that is rapid beat-to-beat heterometric adaptations (Starling’s law of the heart) and otherwise progressive structural and inotropic homeometric adaptations (Anrep’s law of the heart) to changes in loading conditions [61]. Thus the RV adaptation to pulmonary hypertension is homeometric with increased contractility, eventual hypertrophy and preserved dimensions. Failure of this mechanism depending on rate of onset and magnitude of increase in Ppa requires heterometric adaptation, with increased RV dimensions [60, 61, 62••].
Accordingly, RV failure can be defined as a dyspnea fatigue syndrome with eventual systemic congestion caused by the insufficient adaptation of systolic function (homeometric adaptation, Anrep) to increased afterload and involvement of increased dimensions (heterometric adaptation, Starling) to maintain RV flow output adapted to metabolic demand.
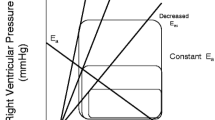
The evaluation of the adequacy of RV-arterial coupling requires a quantification of RV afterload. There are several equally valid estimations of RV afterload [62••]. The first is maximum wall tension, which is however unpractical because of the irregular shape of the RV and regional inhomogeneous contraction. The second is hydraulic power, calculated from the integration of pressure and flow waves, thus integrating its oscillatory component. Because of the near-constancy of RC-time, Wosc can be approximated to 23 % of Wtot, or 1.3 times mean power in all circumstances [54]. The third is arterial elastance (Ea) measured on a RV pressure–volume loop, corresponding to a measurement of afterload as it is “seen” by the ventricle. The RV pressure–volume loop also allows to identify a point of maximal elastance (Emax), which is the gold standard measure of load-independent contractility in an intact heart, and thus the calculation of an Emax/Ea ratio as a measurement of the coupling of ventricular to arterial function.
Mathematical modeling shows that the optimal matching of systolic ventricular and arterial elastances occurs at an Emax/Ea ratio around 1.5.
However, the complex geometry of the RV makes functional evaluations with measurement of instantaneous volume changes technically difficult, and the determination of Emax may be unreliable because of the particular shape of the RV pressure–volume loop and non-coincidence of end-ejection and end-systole. This problem can be overcome by measuring pressure–volume loops at several levels of preload [63]. On the other hand, a single-beat method to calculate Emax and Ea from instanteous ventricular pressure and flow output measurements has been validated for the RV [64]. The approach has been implemented in experimental animal studies to show for example that acutely administered prostacyclin has no intrinsic inotropic effect [65], and that β-blocker agents which deteriorate RV-arterial coupling acutely [64] may improve RV-arterial coupling chronically because of an improvement in contractility [66].
The coupling of RV function to the pulmonary circulation has been reported in patients with pulmonary arterial hypertension (PAH) with single-beat calculation of the Emax/Ea ratio from magnetic resonance imaging (MRI) volume and right heart catheter pressure measurements [67]. As compared to controls, Emax was almost doubled, but Emax/Ea was decreased, indicating insufficient homeometric adaptation and pending RV failure. These results were confirmed by Emax/Ea ratio measurements using conductance catheters and the Valsalva manoeuvre to decrease venous return [68•]. In that study, Emax/Ea was maintained in the patients with idiopathic PAH, but decreased in systemic sclerosis-associated PAH [68•], in keeping with previous similar conclusions reached on the basis of an analysis of RV-arterial coupling by plotting pressure as a function of stroke volume, or so-called the “pump function graph” [62••, 69].
Since Emax/Ea can be simplified to a ratio of volumes, MRI has also been recently implemented for the estimation of RV-arterial coupling estimated by the ratio of stroke volume (SV) to end-systolic volume (ESV). In a study on 139 patients referred for pulmonary hypertension, RV “Emax/Ea” estimated by the volume ratio was shown to decrease progressively with increasing severity of pulmonary hypertension [70]. The SV/ESV ratio contains the same functional information as an ejection fraction, but with less preload-dependency. The clinical relevance of the SV/ESV as measure of RV-arterial coupling is being evaluated.
A derived simplified measure of the adequacy of RV systolic function adaptation to afterload is contractile reserve, defined by the increase in RV systolic pressure during an exercise stress. Grunig recently reported that RV contractile reserve is an important predictor of survival in patients with severe pulmonary hypertension, with a cut-off value of 30 mmHg [71••].
Conclusions
Evaluation of pulmonary hypertension requires understanding of normal pulmonary vascular pressure–flow relationships and right ventriculo-arterial coupling. Recent progresses in imaging combined with gold standard physiological concepts have made possible bedside measurements of the pulmonary circulation and right ventricular function for improved detection, diagnosis, follow-up and prognostication of patients with pulmonary vascular diseases.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• West JB. The role of the fragility of the pulmonary blood-gas barrier in the evolution of the pulmonary circulation. Am J Physiol Regul Integr Comp Physiol. 2013;304:R171–6. Enlightening review allowing for the understanding why the pulmonary circulation in mammals is a separate high flow-low pressure vascular system.
Swan HJC, Ganz W, Forrester JS, et al. Catheterization of the heart in man with use of a flow-directed catheter. N Engl J Med. 1970;283:447–51.
Howell JBL, Permutt S, Proctor DF, Riley RL. Effect of inflation of the lung on different parts of the pulmonary vascular bed. J Appl Physiol. 1961;16:71–6.
• Naeije R, Boerrigter BG. Pulmonary hypertension at exercise in COPD: does it matter? Eur Respir J. 2013;41:1002–4. Brief review with pressure recordings showing the influence of altered respiratory mechanics of pulmonary vascular pressure tracings in patients with COPD.
Halpern SD, Taichman DB. Misclassification of pulmonary hypertension due to reliance on pulmonary capillary wedge pressure rather than left ventricular end-diastolic pressure. Chest. 2009;136:37–43.
Battacharya J, Nanjo S, Staub NC. Micropuncture measurement of lung microvascular pressure during 5-HT infusion. J Appl Physiol. 1982;52:634–7.
Cope DK, Grimbert F, Downey JM, Taylor AE. Pulmonary capillary pressure: a review. Crit Care Med. 1992;20:1043–56.
Gaar Jr KA, Taylor AE, Owens LJ, Guyton AC. Pulmonary capillary pressure and filtration coefficient in the isolated perfused lung. Am J Physiol. 1967;213:910–4.
Fesler P, Pagnamenta A, Vachiéry JL, et al. Single arterial occlusion to locate resistance in patients with pulmonary hypertension. Eur Respir J. 2003;21:31–6.
Kim NH, Fesler P, Channick RN, et al. Preoperative partitioning of pulmonary vascular resistance correlates with early outcome after thromboendarterectomy for chronic thromboembolic pulmonary hypertension. Circulation. 2004;109:18–22.
• Toshner M, Suntharalingam J, Fesler P, et al. Occlusion pressure analysis role in partitioning of pulmonary vascular resistance in CTEPH. Eur Respir J. 2012;40:612–7. Bold attempt at an interesting clinical application of the single occlusion method for the diagnosis of proximal operable CTEPH.
Maggiorini M, Mélot C, Pierre S, et al. High altitude pulmonary edema is initially caused by an increased capillary pressure. Circulation. 2001;103:2078–83.
Naeije R, Mélot C, Mols P, Hallemans R. Effects of vasodilators on hypoxic pulmonary vasoconstriction in normal man. Chest. 1982;82:404–10.
Naeije R, Mélot C, Niset G, Delcroix M, Wagner PD. Improved arterial oxygenation by a pharmacological increase in chemosensitivity during hypoxic exercise in normal subjects. J Appl Physiol. 1993;74:1666–71.
Mélot C, Naeije R, Hallemans R, Lejeune P, Mols P. Hypoxic pulmonary vasoconstriction and pulmonary gas exchange in normal man. Respir Physiol. 1987;68:11–27.
Kovacs G, Berghold A, Scheid S, Olschewski H. Pulmonary artery pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34:888–94.
Granath A, Jonsson B, Strandell T. Circulation in healthy old men, studied by right heart catheterization at rest and during exercise in supine and sitting position. Acta Med Scand. 1964;176:425–46.
Granath A, Strandell T. Relationships between cardiac output, stroke volume, and intracardiac pressures at rest and during exercise in supine position and some anthropometric data in healthy old men. Acta Med Scand. 1964;176:447–66.
•• Naeije R, Chesler NC. Pulmonary circulation at exercise. Compr Physiol. 2012;2:711–41. Comprehensive actualized review of the pulmonary circulation at exercise.
Permutt S, Bromberger-Barnea B, Bane HN. Alveolar pressure, pulmonary venous pressure and the vascular waterfall. Med Thorac. 1962;19:239–60.
West JB, Dollery CT, Naimark A. Distribution of blood flow in isolated lung: relation to vascular and alveolar pressures. J Appl Physiol. 1964;19:713–24.
Leeman M, Lejeune P, Closset J, Vachiéry JL, Mélot C, Naeije R. Nature of pulmonary hypertension in canine oleic acid pulmonary edema. J Appl Physiol. 1990;69:293–8.
Zapol WM, Snider MT. Pulmonary hypertension in severe acute respiratory failure. N Engl J Med. 1977;296:476–80.
Naeije R, Lipski A, Abramowicz M, et al. Nature of pulmonary hypertension in congestive heart failure. Effects of cardiac transplantation. Am J Respir Crit Care Med. 1997;147:881–7.
Linehan JH, Haworth ST, Nelin LD, Krenz GS, Dawson CA. A simple distensible model for interpreting pulmonary vascular pressure-flow curves. J Appl Physiol. 1992;73:987–94.
Krenz GS, Dawson CA. Flow and pressure distributions in vascular networks consisting of distensible vessels. Am J Physiol Heart Circ. 2003;284:H2192–203.
Reeves JT, Linehan JH, Stenmark KR. Distensibility of the normal human lung circulation during exercise. Am J Physiol Lung Cell Mol Physiol. 2005;288:L419–25.
Argiento P, Chesler N, Mulè M, et al. Exercise stress echocardiography for the study of the pulmonary circulation. Eur Respir J. 2010;35:1273–8.
Argiento P, Vanderpool RR, Mule M, et al. Exercise stress echocardiography of the pulmonary circulkation: limits of normal and sex differences. Chest. 2012;142:1158–65.
Lalande S, Yerly P, Faoro V, Naeije R. Pulmonary vascular distensibility predicts aerobic capacity in healthy individuals. J Physiol. 2012;590:4279–88.
Groepenhoff H, Overbeek MJ, Mulè M, et al. Exercise pathophysiology in patients with chronic mountain ickness. Chest. 2012;142:877–84.
•• Naeije R, Vanderpool R, Dhakal B, et al. Exercise-induced pulmonary hypertension: physiological basis and methodological concerns. Am J Respir Crit Care Med. 2013;187:576–83. Actualized comprehensive review on the diagnosis and mechanisms of exercise-induced pulmonary hypertension.
Pavelescu A, Vanderpool R, Vachiéry JL, Grunig E, Naeije R. Echocardiography of pulmonary vascular function in asymptomatic carriers of BMPR2 mutations. Eur Respir J. 2012;40:1287–9.
Mélot C, Delcroix M, Closset J, et al. Starling resistor vs. distensible vessel models for embolic pulmonary hypertension. Am J Physiol. 1995;268:H817–27.
Lau E. Early detection of pulmonary vascular disease in pulmonary arterial hypertension: time to move forward. Eur Heart J. 2011;32:2489–98.
Whittaker SRF, Winton FR. The apparent viscosity of blood flowing in the isolated hindlimb of the dog, and its variation with corpuscular concentration. J Physiol. 1933;78:339–69.
Naeije R, Vanderpool R. Pulmonary hypertension in chronic mountain sickness. High Alt Med Biol. 2013;14:117–25.
Faoro V, Huez S, Vanderpool RR, et al. Pulmonary circulation and gas exchange at exerciuse in Sherpas at high altitude. J Appl Physiol. 2013.
Moraes DL, Colucci WS, Givertz MM. Secondary pulmonary hypertension in chronic heart failure. The role of endothelium in pathophysiology and management. Circulation. 2000;102:1718–23.
Hoeper MM, Barberà JA, Channick RN, et al. Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S85–96.
•• Naeije R, Vachiery J, Yerly P, Vanderpool R. The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J. 2013;41:217–23. Refreshed view on how to interprete pulmonary vascular pressure gradients in the differential diagnosis of pulmonary hypertension associated with left heart conditions.
Harvey RM, Enson Y, Ferrer MI. A reconsideration of the origins of pulmonary hypertension. Chest. 1971;59:82–94.
Gerges C, Gerges M, Lang MB, et al. Diastolic pulmonary vascular pressure gradient: a predictor of prognosis in "out-of-proportion" pulmonary hypertension. Chest. 2013;143:758–66.
Reeves JT, Dempsey JA, Grover RF. Pulmonary circulation during exercise. In: Weir EK, Reeves JT, editors. Pulmonary vascular physiology and physiopathology. Chap 4. New York: Marcel Dekker; 1989. p. 107–33.
Kovacs G, Olschewski A, Berhold A, Oschewski H. Pulmonary vascular resistance during exercise in normal subjects: a systematic review. Eur Respir J. 2012;39:319–28.
Stickland MK, Welsh RC, Petersen SR, et al. Does fitness level modulate the cardiovascular hemodynamic response to exercise? J Appl Physiol. 2006;100:1895–901.
Bergel DH, Milnor WR. Pulmonary vascular impedance in the dog. Circ Res. 1966;16:401–15.
Milnor WR, Bergel DH, Bargainer JD. Hydraulic power associated with pulmonary blood flow and its relation to heart rate. Circ Res. 1966;19:467–80.
Lankhaar JW, Westerhof N, Faes TJ, et al. Quantification of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2006;291:H1731–7.
Lankhaar JW, Westerhof N, Faes TJ, et al. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J. 2008;29:1688–95.
Saouti N, Westerhof N, Helderman F, et al. RC time constant of single lung equals that of both lungs together: a study in chronic thromboembolic pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2009;297:H2154–60.
Reuben SR. Compliance of the pulmonary arterial system in disease. Circ Res. 1971;29:40–50.
Bonderman D, Martischnig AM, Vonbank K, et al. Right ventricular load at exercise is a cause of persistent exercise limitation in patients with normal resting pulmonary vascular resistance after pulmonary endarterectomy. Chest. 2011;139:122–7.
Saouti N, Westerhof N, Helderman F, et al. Right ventricular oscillatory power is a constant fraction of total power irrespective of pulmonary artery pressure. Am J Respir Crit Care Med. 2010;182:1315–20.
• Tedford RJ, Hassoun PM, Mathai SC, et al. Pulmonary capillary wedge pressure augments right ventricular pulsatile loading. Circulation. 2012;125:289–97. Study on a large patient population reporting on the time constant in the pulmonary versus systemic circulations and the effects of heart failure.
Pagnamenta A, Vanderpool R, Brimioulle S, Naeije R. Proximal pulmonary arterial obstruction decreases the time constant of the pulmonary circulation and increases right ventricular afterload. J Appl Physiol. 2013;114:1586–92.
• MacKenzie Ross RV, Toshner MR, Soon E, Naeije R, Pepke-Zaba J. Decreased time constant of the pulmonary circulation in chronic thromboembolic pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2013;305:H259–64. Clinical study showing that proximal CTEPH is associated with a decreased time constant of the pulmonary circulation.
Chemla D, Castelain V, Provencher S, Humbert M, Simonneau G, Hervé P. Evaluation of various empirical formulas for estimating mean pulmonary artery pressure by using systolic pulmonary artery pressure in adults. Chest. 2009;135:760–8.
Yock P, Popp R. Noninvasive estimation of right ventricular systolic pressure by Doppler ultrasound in patients with tricuspid regurgitation. Circulation. 1984;70:657–62.
Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation. 2009;120:992–1007.
Sagawa K, Maughan L, Suga H, Sunagawa K. Cardiac contraction and the pressure–volume relationship. New York: Oxford University Press; 1988.
•• Vonk Noordegraaf A, Westerhof N. Describing right ventricular function. Eur Respir J. 2013;41:1419–23. Concise piece essential to the modern understanding of right ventricular function.
Maughan WL, Shoukas AA, Sagawa K, Weisfeldt ML. Instantaneous pressure-volume relationship of the canine right ventricle. Circ Res. 1979;44:309–15.
Brimioulle S, Wauthy P, Ewalenko P, et al. Single-beat estimation of right ventricular end-systolic pressure-volume relationship. Am J Physiol Heart Circ Physiol. 2003;284:H1625–30.
Kerbaul F, Brimioulle S, Rondelet B, Dewachter C, Hubloue I, Naeije R. How prostacyclin improves cardiac output in right heart failure in conjunction with pulmonary hypertension. Am J Respir Crit Care Med. 2007;175:846–50.
De Man FS, Handoko ML, van Ballegoij LL, et al. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012;5:97–105.
Kuehne T, Yilmaz S, Steendijk P, et al. Magnetic resonance imaging analysis of right ventricular pressure-volume loops: in vivo validation and clinical application in patients with pulmonary hypertension. Circulation. 2004;110:2010–6.
• Tedford RJ, Mudd JO, Girgis RE, et al. Right ventricular dysfunction in systemic sclerosis associated pulmonary arterial hypertension. Circ Heart Fail. 2013. Important paper on RV-arterial coupling in idiopathic versus scleroderma-associated pulmonary hypertension.
Overbeek MJ, Lankhaar JW, Westerhof N, et al. Right ventricular contractility in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. Eur Respir J. 2008;31:1160–6.
Sanz J, García-Alvarez A, Fernández-Friera L, et al. Right ventriculo-arterial coupling in pulmonary hypertension: a magnetic resonance study. Heart. 2012;98:238–43.
•• Grunig E, Tiede H, Enyiamayew EO, et al. patients with pulmonary arterial hypertension. Circulation. 2013. Timely paper drawing attention on the importance of right ventricular contractile reserve in severe pulmonary hypertension.
Compliance with Ethics Guidelines
Conflict of Interest
Robert Naeije declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Naeije, R. Physiology of the Pulmonary Circulation and the Right Heart. Curr Hypertens Rep 15, 623–631 (2013). https://doi.org/10.1007/s11906-013-0396-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-013-0396-6