
Abstract
Pheochromocytomas and paragangliomas are highly heterogeneous neuroendocrine tumors that must be considered not only in patients with hypertension and other manifestations of catecholamine excess but also in patients with incidentalomas or mutations in one of the ten tumor susceptibility genes identified to date. To first think of the tumor remains the critical step for screening in patients with signs and symptoms. In these patients, biochemical testing is straightforward and should include measurements of plasma or urinary metanephrines, comprising separately measured normetanephrine and metanephrine. Tumors due to an underlying germline mutation are often found in the absence of hypertension or other signs or symptoms of the tumor. Screening for disease in these patients can benefit from an individualized approach according to the particular mutation. Additional measurements of methoxytyramine, the metabolite of dopamine, can be useful in patients with mutations of succinate dehydrogenase genes or patients who are at risk for malignancy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pheochromocytomas are a rare, usually curable cause of secondary hypertension. About 85% arise from chromaffin cells of the adrenal medulla. The others, referred to as “paragangliomas,” arise from extra-adrenal chromaffin tissue. Paragangliomas form mainly in the abdomen but also can occur at numerous other extra-adrenal sites such as in the urinary bladder, mediastinum, and head and neck regions. Head and neck paragangliomas include glomus tumors, chemodectomas, carotid body tumors, and jugulotympanic tumors. Unlike abdominal and thoracic paragangliomas, these tumors arise from parasympathetic-associated tissue and usually do not produce catecholamines or cause hypertension, so they are not considered further in this article.
The blood pressure disturbances and most of the other diverse signs and symptoms of pheochromocytomas and abdominal or thoracic paragangliomas result primarily from tumoral production and secretion of catecholamines. Consequently, patients with high blood pressure and symptoms of catecholamine excess are those in whom the tumors are most frequently suspected and screened for. Among patients with hypertension at general outpatient clinics, the prevalence of pheochromocytoma is about 0.2% to 0.6% [1–3], but it may be as high as 4% in patients with refractory hypertension [4].
Because of the high prevalence of hypertension, the rarity of the tumors, and the nonspecific and highly variable nature of the symptoms of catecholamine excess, pheochromocytomas and paragangliomas are frequently searched for but only occasionally found. More often, the tumors remain unsuspected. In such patients, catecholamine secretion can lead to a very sudden appearance of clinical manifestations and lethal complications. Thus, as revealed by autopsy studies, the tumors are frequently missed, usually contributing to premature death [5–7]. These studies suggest that the true prevalence of chromaffin cell tumors may be as high as 1:1,000 and is not accurately reflected by the tumors diagnosed during life.
Although it is commonly reported that 80% to 90% of patients with pheochromocytoma have hypertension, many patients are now being described who are completely normotensive and asymptomatic when the tumors are found. This change likely reflects the growing proportion of patients diagnosed with pheochromocytoma or paraganglioma as a consequence of routine screening because of underlying hereditary syndromes or because a mass is discovered incidentally during imaging studies for unrelated conditions. Among such patients, up to 50% or more are normotensive [8, 9]. These observations demonstrate that hypertension is not always a hallmark feature of pheochromocytoma, partly explaining why many of these tumors remain unsuspected and undetected throughout life.
Initial Biochemical Testing
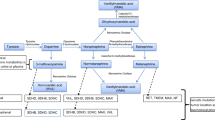
Advances in understanding catecholamine metabolism have led to a paradigm shift in the biochemical diagnosis of pheochromocytoma, away from measurements of catecholamines to a focus on their O-methylated metabolites [10–12]. This shift has followed several observations, including findings that adrenal medullary cells and pheochromocytoma tumor cells contain catechol-O-methyltransferase (COMT), the enzyme that converts norepinephrine to normetanephrine, epinephrine to metanephrine, and dopamine to methoxytyramine [13] (Fig. 1). Thus, the adrenal glands, not the more commonly considered liver and kidneys, represent the single largest site of catecholamine O-methylation, accounting for at least 90% of all circulating metanephrine and 23% of normetanephrine [14].

Pathways of catecholamine metabolism by monoamine oxidase (MAO) within sympathetic nerves and by catechol-O-methyltransferase (COMT) within adrenalmedullary cells. Norepinephrine (NE) in sympathetic nerves is deaminated to 3,4-dihydroxyphenylglycol (DHPG), which is then further O-methylated in extraneuronal tissues to 3-methoxy-4-hydroxyphenylglycol (MHPG). In adrenalmedullary cells, norepinephrine is either O-methylated to normetanephrine (NMN) or N-methylated by phenylethanolamine-N-methyltransferase (PNMT) to epinephrine (EPI), which is O-methylated to metanephrine (MN). Note that substantial proportions of circulating normetanephrine and metanephrine are derived from metabolism within adrenalmedullary cells and that most initial metabolism of norepinephrine and epinephrine occurs because of leakage of catecholamines from storage vesicles into the cytoplasm. This is a continuous process that is independent of variations in catecholamine release
Normally the O-methylation pathway represents a minor route of catecholamine metabolism; deamination of norepinephrine within sympathetic nerves is the major pathway. Intraneuronal deamination is followed by O-methylation of the deaminated metabolite in extraneuronal tissues and finally oxidation in the liver to vanillylmandelic acid (VMA), the major urinary metabolite of norepinephrine and epinephrine.
In patients with pheochromocytoma, intratumoral O-methylation becomes a dominant pathway of catecholamine metabolism. Consequently, the presence of the tumor leads to relatively large increases in production of the O-methylated metabolites, compared with minor increases of deaminated metabolites. Because of the continuous high rate of intratumoral catecholamine O-methylation, and because some tumors secrete catecholamines episodically or in low amounts, patients with pheochromocytoma usually have relatively larger and more consistent increases of plasma normetanephrine or metanephrine than of the parent catecholamines [15].
The particularly high diagnostic sensitivity of measurements of plasma free metanephrines has now been confirmed by numerous independent studies [16–20, 21•] (Table 1). In one of the largest series [17], involving over 200 patients with pheochromocytoma and more than 600 patients in whom the tumor was excluded, measurements of plasma free metanephrines provided the most sensitive diagnostic test, urinary and plasma catecholamines offered intermediate sensitivity, and urinary total metanephrines and VMA were the least sensitive.
Measurements of urinary fractionated metanephrines also offer relatively high diagnostic sensitivity for screening chromaffin cell tumors [17, 22]. These measurements are usually performed after a deconjugation step, however, and reflect mainly sulfate-conjugated metabolites, which are principally cleared by the kidneys and represent the major forms excreted in urine [23]. These sulfate-conjugated metabolites are formed from the free metabolites by an enzyme present in high concentrations in digestive tissues, which functions to inactivate dietary amines and conjugate the large amounts of norepinephrine, dopamine, and their metabolites produced locally [24].
The dietary and local mesenteric organ sources of sulfate conjugates dilute the contribution of chromaffin tissue sources to the sulfate conjugates. Consequently, sulfate conjugated metanephrines in urine and plasma offer slightly lower diagnostic sensitivity than the free metabolites and also suffer from influences of metabolism of dietary and locally produced amines within the gut [17, 25, 26•]. Thus, receiver-operating characteristic curves indicate that at equivalent levels of specificity, the sensitivity of plasma free metanephrines is higher than that of urinary fractionated metanephrines, whereas at equivalent levels of sensitivity, the specificity of plasma free metanephrines is higher than that of all other tests, including urinary fractionated deconjugated metanephrines [17]. Thus, at similar levels of high diagnostic sensitivity, tests of plasma free metanephrines exclude pheochromocytoma in more patients without the tumor than do tests of urinary fractionated metanephrines. False-positive rates for the latter test have been reported to be particularly high [27•].
With these findings in mind, initial biochemical testing for pheochromocytoma ideally should use measurements of plasma free metanephrines as the principal test, but the low concentrations of free metanephrines in plasma are not as easy to measure as the much higher concentrations of deconjugated metanephrines in urine. The metabolites may be measured by immunoassay or by liquid chromatography followed by either electrochemical (LC-EC) or tandem mass spectrometric detection (LC-MS/MS) [10, 28•]. As shown by interlaboratory proficiency programs, not all measurement methods provide optimal results. Immunoassays in particular appear to suffer from unacceptable error, which may negate all gains achieved by measurement of a biomarker produced within tumor tissue [28•, 29, 30•]. In such situations, accurate measurements of urinary fractionated metanephrines by a high-pressure (HP)LC-EC or LC-MS/MS method may be preferable to inaccurate measurements of plasma free metanephrines by immunoassay.
There are also preanalytical considerations. Blood sampling for measurements of plasma free metanephrines should be carried out after 30 min of supine rest. Sampling in the seated position, though easier and more practical for phlebotomists, is associated with at least 30% higher plasma concentrations of free metanephrines in subjects without pheochromocytoma [26•, 31]. The result can be a large increase in the number of false-positive results, particularly when reference intervals are established correctly in blood samples taken in the supine position.
With these issues in mind, the best initial test for biochemical diagnosis remains controversial, with some clinicians maintaining a preference for urinary fractionated metanephrines [32]. Nevertheless, there is now general consensus that initial testing for pheochromocytoma should include either measurements of plasma free or urinary fractionated metanephrines [11, 12, 33].
When urinary fractionated metanephrines are measured, it may be useful to combine the test with additional measurements of urinary catecholamines [18, 32]. However, when plasma free metanephrines are measured by an accurate LC-EC or LC-MS/MS method, there is no need for additional measurements of catecholamines, which are likely only to increase the numbers of false-positive results and are unlikely to lead to the detection of additional tumors not indicated by elevated levels of normetanephrine and metanephrine [17].
Follow-up Biochemical Testing
If measurements are accurate and appropriate reference intervals are used, findings of normal plasma concentrations of metanephrines effectively rule out pheochromocytoma in the hypertensive and symptomatic patient, so that no further testing is required. Thus, follow-up testing usually is necessary only for patients with a positive result [34]. The nature of follow-up testing depends on the likelihood that the positive result indicates a tumor, which is best assessed from the extent of increase of the test result. Increases in plasma concentrations of normetanephrine above 400 ng/L (2.2 nmol/L) or of metanephrine above 236 ng/L (1.2 nmol/L) are extremely rare in patients without pheochromocytoma but occur in about 80% of patients with the tumor [34]. Assuming there was no laboratory error and provided that samples were not taken during a hypertensive crisis or an acute, severe medical emergency (e.g., in the emergency room, during shock, hypoglycemia, respiratory distress), the likelihood of pheochromocytoma in such patients is so high that the immediate task is to locate the tumor.
The remaining problem is to confirm or exclude pheochromocytoma in patients with positive results in the equivocal range. Because of the rarity of the tumor, false-positive results in this range can be expected to outnumber true-positive results [27•]. Therefore the first consideration should be possible sources of false-positive results, including the clinical condition of the patient, inappropriate sampling conditions, laboratory error, and medications likely to interfere with analytic results or to increase levels of normetanephrine or metanephrine.
The clinical condition of the patient can be of paramount importance in the critical care setting or in patients with conditions known to increase activity of the sympathetic nervous system and plasma concentrations of catecholamines and their metabolites [35]. If the initial blood sample was taken in a less than ideal situation with the patient in the seated position or under stress, then repeat sampling is called for [31].
The clonidine-suppression test can be particularly useful in patients with consistently elevated plasma concentrations of normetanephrine, with or without elevations of plasma norepinephrine. Initially introduced by Bravo and colleagues [36], the test was first used to distinguish tumoral from sympathoneuronal sources of elevated plasma norepinephrine. Clonidine-induced falls in plasma norepinephrine can occur in patients with pheochromocytoma who have normal or mildly elevated levels of norepinephrine, however [34]. In such patients, only small amounts of circulating norepinephrine are derived from the tumor; most originates from sympathetic nerves and is responsive to clonidine. This problem of less than ideal diagnostic sensitivity with the use of norepinephrine as a diagnostic end point is largely overcome by measurements of plasma normetanephrine [34].
Screening for Hereditary Pheochromocytoma and Paraganglioma
Most pheochromocytomas are sporadic, but a significant proportion result from germ-line mutations of several tumor susceptibility genes (Table 2). Mutations of the rearranged during transfection (RET) gene in multiple endocrine neoplasia type 2 (MEN 2), of the von Hippel-Lindau (VHL) gene in VHL syndrome, of the neurofibromatosis type 1 (NF1) gene in von Recklinghausen disease, and of genes encoding succinate dehydrogenase (SDH) subunits B (SDHB) and D (SDHD) are the best-known causes of hereditary pheochromocytomas and paragangliomas. Mutations of the gene for SDH subunit C (SDHC) are a less frequent cause of catecholamine-producing pheochromocytomas and paragangliomas [37]. Mutations of genes encoding the SDH complex assembly factor 2 (SDHAF2), transmembrane protein 127 (TMEM127), SDH subunit A (SDHA), and MYC associated factor X (MAX) have been more recently identified as further hereditary causes of the tumors [38•, 39••, 40, 41••]. Altogether, ten tumor susceptibility genes are now recognized to be responsible for hereditary chromaffin cell tumors.
Reported frequencies of germline mutations of VHL, RET, SDHD, and SDHB genes among patients with chromaffin cell tumors range from 27% to 32% [42–44]. These studies did not take into account all known germ-line mutations, however. As more are identified, the contribution of hereditary factors is likely to increase further. Even so, it is clear from the data at hand that pheochromocytomas and paragangliomas have one of the most pronounced hereditary components among all neoplastic disorders.
Among all patients with NF1 and most with VHL syndrome and MEN 2, the hereditary basis is clear from the syndromic nature of the disorder, often with a positive family history. The common clinical manifestations on which the diagnosis of NF1 is made include café au lait spots, neurofibromas, axillary or inguinal freckling, optic glioma, Lisch nodules, and osseous lesions [45]. The penetrance of pheochromocytoma in NF1 is relatively low (<2%), and unlike other hereditary conditions, it occurs usually later in life [46]. Apart from pheochromocytomas, patients with VHL syndrome are at additional risk for renal carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts. Among patients with MEN 2, the penetrance of medullary thyroid carcinoma is particularly high; parathyroid neoplasia further characterizes patients with MEN 2A and multiple mucosal neuromas and a marfanoid habitus characterize those with MEN 2B [47].
These clinical features make diagnosis of the underlying germ-line mutation relatively easy in most patients with NF1, VHL syndrome, and MEN 2. Nevertheless, as shown in one study, the presence of a VHL or RET mutation can sometimes be less clear, and as many as 24% of patients with apparently nonsyndromic pheochromocytoma may have an underlying mutation of either VHL, RET, SDHD, or SDHB genes [48]. Other studies have indicated lower proportions, however (12–19%), with most germ-line mutations among patients with apparently sporadic pheochromocytoma restricted to SDHD and SDHB genes [43, 44, 49]. Lack of a distinct syndromic presentation combined with low penetrance of disease and unclear family history in patients with these mutations has led to suggestions that all patients with chromaffin cell tumors should be considered for genetic testing. However, at an international meeting convened to develop guidelines on this and other issues, it was recommended that despite a reasonable argument for more widespread genetic testing, it is neither appropriate nor currently cost-effective to test every disease-causing gene in every patient with a tumor; rather, the decision to test and which genes to test requires judicious consideration of numerous factors [11].
An early age of disease presentation is commonly cited as one of the most important factors that should dictate whether to test for tumor susceptibility genes; other evidence indicates that testing of SDHD, SDHB, and VHL genes is particularly warranted in patients presenting with norepinephrine-producing tumors before the age of 35 years [46]. Tumor location is also important (Table 2). Extra-adrenal tumors are relatively common in patients with SDHD and SDHB mutations, with head and neck paragangliomas particularly common in patients with SDHD mutations [50]; thus, testing of these genes is especially warranted in patients with tumors at these locations. SDHB mutations carry a particularly high risk for malignant disease, mandating testing of this gene in all patients with evidence of metastases.
Mutations of the various tumor susceptibility genes also give rise to distinct catecholamine metabolomic and secretory signatures (Table 2), which can be ascertained during biochemical testing and can also provide clues to underlying gene mutations [51•]. Tumors in patients with MEN 2 and NF1 are almost always confined to the adrenals and are characterized by increases in plasma free metanephrine, indicating epinephrine production. In contrast, relative absence of epinephrine production characterizes tumors in patients with VHL, SDHD, and SDHB mutations, including those at adrenal locations. Finally, increases in methoxytyramine, the metabolite of dopamine, characterize up to 70% of patients with mutations of SDHD and SDHB genes.
The distinct mutation-dependent biochemical profiles (Table 2) not only are useful for stratifying patients for genetic testing but also are important for routine screening of tumors in patients with identified mutations. For mutations conferring a high risk of disease, such screening is generally recommended at yearly intervals and should include biochemical testing for evidence of excess catecholamine production. Because tumors in patients with VHL mutations are characterized by solitary increases in normetanephrine, emphasis should be directed at measurements of this metabolite; solitary small increases of metanephrine are likely to reflect false positives. In contrast, for patients with RET and NF1 mutations, emphasis should be placed on measurements of both normetanephrine and metanephrine, as both metabolites invariably show increases; nevertheless, solitary increases in metanephrine cannot be ignored. Although measurements of plasma free methoxytyramine are not called for during screening of most sporadic and familial pheochromocytomas, these measurements are particularly important for screening of tumors in patients with SDHB and SDHD mutations, some of which can be diagnosed only by solitary increases in that metabolite.
Screening for Malignant or Recurrent Disease
There are currently no reliable histopathologic methods to distinguish benign from malignant pheochromocytoma; only the presence of metastases at sites where no chromaffin tissue should be expected (bones, liver, lungs, and lymph nodes) establishes a definitive diagnosis of malignant pheochromocytoma [52]. Although there are no markers to reliably predict the development of malignant pheochromocytoma, several factors are associated with increased risk of malignancy. Rates of malignancy are 3.6-fold higher in extra-adrenal tumors than adrenal tumors [53••], and there are now increasing indications that large tumor size is associated with malignancy [53••, 54–59]. Tumors in patients with mutations of the SDHB gene have a particularly high rate of malignancy [60, 61], reflecting both the typically extra-adrenal location and large size reached by the tumors in these patients.
Extra-adrenal location, large size, and presence of SDHB mutations are all important to consider when screening for metastatic disease in a patient with diagnosed pheochromocytoma or paraganglioma. Presence of these risk factors may justify more extensive preoperative imaging to exclude metastases in these patients than in others at lower risk for malignancy. Similarly, such factors may also be used to adjust the intensiveness of routine postoperative screening for malignancy during long-term follow-up. Although the risk of malignancy is quite low, it is nevertheless always important to consider it during the long-term follow-up now recommended for all patients with chromaffin cell tumors. Follow-up also should always consider loco-regional recurrent disease, particularly multifocal disease in patients with mutations of tumor susceptibility genes.
Several studies have indicated associations of malignant chromaffin cell tumors with increased urinary output or plasma concentration of dopamine [55, 62–64]. In part, this finding appears to reflect associations of malignancy with SDHB mutations and extra-adrenal tumors, often characterized by dopamine production. More recently, elevated plasma concentrations of methoxytyramine have been shown to offer more sensitivity to indicate tumoral dopamine production and likelihood of malignancy [53••]. This study also indicated that the association of elevations of methoxytyramine with malignancy was present in patients with adrenal tumors and in those without SDHB mutations.
In addition to screening for paragangliomas in patients with SDHB and SDHD mutations, measurements of methoxytyramine should therefore also be useful for screening for malignant disease, but measurements of methoxytyramine need not be incorporated in screening for all pheochromocytomas and paragangliomas. Most patients with these tumors have normal plasma concentrations of methoxytyramine, and unselected general testing with a combination of normetanephrine, metanephrine, and methoxytyramine is only likely to confuse diagnostic decision-making because of increased numbers of false-positive results from dietary dopamine [26•]. Measurements of plasma methoxytyramine are better restricted to the routine screening of patients with SDHB and SDHD mutations or follow-up for possible metastatic disease in a patient in whom a pheochromocytoma or paraganglioma has already been diagnosed. For such testing, blood must be sampled after an overnight fast [26•].
Conclusions
Initial screening for catecholamine-producing pheochromocytomas and paragangliomas is now widely recommended to always include measurements of urinary or plasma metanephrines or both, these comprising separately measured normetanephrine and metanephrine. Measurements of plasma free metanephrines in particular provide diagnostic sensitivity exceeding 96% with specificity of 85% to 100%, but they are not always available or offered for measurement using suitably accurate and precise liquid chromatography–based methods. In such situations, measurements of urinary fractionated metanephrines offer an alternative approach with diagnostic sensitivity nearly as good.
Pheochromocytomas and paragangliomas are now recognized as highly heterogeneous neoplasms with ages of onset, secretory profiles, locations, and potentials for malignancy that differ according to underlying genetic mutations. The need to distinguish metastatic from benign tumors and to consider a possible hereditary basis of the disease has led to identification of additional screening methods involving differences in profiles of O-methylated metabolites to stratify patients according to risk of malignant disease or of a particular underlying mutation. Additional measurements of methoxytyramine, the metabolite of dopamine, can be useful to screen for tumors in patients with SDHB and SDHD mutations or to identify the presence of malignancy, but these measurements are not otherwise recommended for general screening.
Patients and family members with identified mutations require an individualized approach to management that includes consideration of distinct patterns of biochemical test results during recommended annual screenings. Because of the increased complexity of appropriately managing patients with chromaffin cell tumors or those who are at risk for the tumors or the development of metastases, the role of referral centers with specialized expertise and technology is becoming increasing important.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Anderson Jr GH, Blakeman N, Streeten DH. The effect of age on prevalence of secondary forms of hypertension in 4429 consecutively referred patients. J Hypertens. 1994;12(5):609–15.
Ariton M, Juan CS, AvRuskin TW. Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocr Pract. 2000;6(3):249–52.
Omura M, Saito J, Yamaguchi K, et al. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27(3):193–202.
Martell N, Rodriguez-Cerrillo M, Grobbee DE, et al. High prevalence of secondary hypertension and insulin resistance in patients with refractory hypertension. Blood Pres. 2003;12(3):149–54.
Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354–60.
McNeil AR, Blok BH, Koelmeyer TD, et al. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust New Zeal J Med. 2000;30(6):648–52.
Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212–5.
Walther MM, Reiter R, Keiser HR, et al. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol. 1999;162(3 Pt 1):659–64.
Mantero F, Terzolo M, Arnaldi G, et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000;85(2):637–44.
Singh RJ. Advances in metanephrine testing for the diagnosis of pheochromocytoma. Clin Lab Med. 2004;24(1):85–103.
Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metabol. 2007;3(2):92–102.
Whiting MJ, Doogue MP. Advances in biochemical screening for phaeochromocytoma using biogenic amines. Clin Biochem Rev. 2009;30(1):3–17.
Eisenhofer G, Keiser H, Friberg P, et al. Plasma metanephrines are markers of pheochromocytoma produced by catechol-O-methyltransferase within tumors. J Clin Endocrinol Metab. 1998;83(6):2175–85.
Eisenhofer G, Rundquist B, Aneman A, et al. Regional release and removal of catecholamines and extraneuronal metabolism to metanephrines. J Clin Endocrinol Metab. 1995;80(10):3009–17.
Eisenhofer G, Huynh TT, Hiroi M, Pacak K. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev Endocr Metab Disord. 2001;2(3):297–311.
Raber W, Raffesberg W, Bischof M, et al. Diagnostic efficacy of unconjugated plasma metanephrines for the detection of pheochromocytoma. Arch Intern Med. 2000;160(19):2957–63.
Lenders JW, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287(11):1427–34.
Sawka AM, Jaeschke R, Singh RJ, Young Jr WF. A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab. 2003;88:553–8.
Unger N, Pitt C, Schmidt IL, et al. Diagnostic value of various biochemical parameters for the diagnosis of pheochromocytoma in patients with adrenal mass. Eur J Endocrinol. 2006;154(3):409–17.
Vaclavik J, Stejskal D, Lacnak B, et al. Free plasma metanephrines as a screening test for pheochromocytoma in low-risk patients. J Hypertens. 2007;25(7):1427–31.
• Hickman PE, Leong M, Chang J, et al. Plasma free metanephrines are superior to urine and plasma catecholamines and urine catecholamine metabolites for the investigation of phaeochromocytoma. Pathology. 2009;41(2):173–7. This study confirms the diagnostic superiority of plasma free metanephrines over other biochemical tests for the diagnosis of pheochromocytoma.
Perry CG, Sawka AM, Singh R, et al. The diagnostic efficacy of urinary fractionated metanephrines measured by tandem mass spectrometry in detection of pheochromocytoma. Clin Endocrinol (Oxf). 2007;66(5):703–8.
Eisenhofer G. Free or total metanephrines for diagnosis of pheochromocytoma: what is the difference? Clin Chem. 2001;47(6):988–9.
Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56(3):331–49.
Lenders JW, Keiser HR, Goldstein DS, et al. Plasma metanephrines in the diagnosis of pheochromocytoma. Ann Intern Med. 1995;123(2):101–9.
• de Jong WH, Eisenhofer G, Post WJ, et al. Dietary influences on plasma and urinary metanephrines: implications for diagnosis of catecholamine-producing tumors. J Clin Endocrinol Metab. 2009;94(8):2841–9. This study established that plasma and urinary methoxytyramine, as well as urinary normetanephrine, were increased by common food sources containing catecholamines. These findings indicate the importance of an overnight fast before sampling of blood for measurements of plasma methoxytyramine and more extended dietary restrictions for urinary measurements.
• Yu R, Wei M. False positive test results for pheochromocytoma from 2000 to 2008. Exp Clin Endocrinol Diabetes. 2010;118(9):577–85. False-positive test results of biochemical testing were examined in this retrospective analysis, which established a particularly high rate for testing of urinary metanephrines, mainly because of physiological influences, effects of medications, and laboratory error.
• Peaston RT, Graham KS, Chambers E, et al. Performance of plasma free metanephrines measured by liquid chromatography-tandem mass spectrometry in the diagnosis of pheochromocytoma. Clin Chim Acta. 2010;411(7–8):546–52. This is an examination of the utility of LC-MS/MS measurements of plasma metanephrines for the diagnosis of pheochromocytoma in comparison to enzyme immunoassay measurements. The study established a useful mass spectrometric method but also showed negative bias of immunoassay measurements with subsequent propensity for false-negative test results.
Pillai D, Ross HA, Kratzsch J, et al. Proficiency test of plasma free and total metanephrines: report from a study group. Clin Chem Lab Med. 2009;47:786–90.
• Pillai D, Callen S. Pilot quality assurance programme for plasma metanephrines. Ann Clin Biochem. 2010;47(Pt 2):137–42. This is a summary of the results of an interlaboratory quality assurance program examining the accuracy and precision of different assay methods for measuring plasma free metanephrines. The analysis indicates limited diagnostic precision of immunoassay measurements. The accuracy data suggest negative bias.
Lenders JW, Willemsen JJ, Eisenhofer G, et al. Is supine rest necessary before blood sampling for plasma metanephrines? Clin Chem. 2007;53(2):352–4.
Young Jr WF. Adrenal causes of hypertension: pheochromocytoma and primary aldosteronism. Rev Endocr Metab Disord. 2007;8(4):309–20.
Grossman A, Pacak K, Sawka A, et al. Biochemical diagnosis and localization of pheochromocytoma: can we reach a consensus? Ann N Y Acad Sci. 2006;1073:332–47.
Eisenhofer G, Goldstein DS, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results. J Clin Endocrinol Metab. 2003;88(6):2656–66.
Eisenhofer G, Friberg P, Pacak K, et al. Plasma metadrenalines: do they provide useful information about sympatho-adrenal function and catecholamine metabolism? Clin Sci (Lond). 1995;88(5):533–42.
Bravo EL, Tarazi RC, Fouad FM, et al. Clonidine-suppression test: a useful aid in the diagnosis of pheochromocytoma. N Engl J Med. 1981;305(11):623–6.
Mannelli M, Ercolino T, Giache V, et al. Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J Med Genet. 2007;44(9):586–7.
• Bayley JP, Kunst HP, Cascon A, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11(4):366–72. This report establishes mutations of the SDHAF2 gene as a novel cause of familial paraganglioma.
•• Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42(3):229–33. This report establishes mutations of the TMEM127 gene as a novel cause of hereditary pheochromocytoma.
Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–20.
•• Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43(7):663–7. This report identifies the tenth pheochromocytoma susceptibility gene.
Bryant J, Farmer J, Kessler LJ, et al. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Canc Inst. 2003;95(16):1196–204.
Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23(34):8812–8.
Mannelli M, Castellano M, Schiavi F, et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab. 2009;94(5):1541–7.
Friedman JM, Birch PH. Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients. Am J Med Genet. 1997;70(2):138–43.
Eisenhofer G, Timmers H, Lenders JW, et al. Age at diagnosis of pheochromocytoma differs according to catecholamine phenotype and tumor location. J Clin Endocrinol Metab. 2011;96:375–84.
Gagel RF. Multiple endocrine neoplasia. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, editors. Williams textbook of endocrinology. 9th ed. Philadelphia: WB Saunders Company; 1998. p. 1627–49.
Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459–66.
Castellano M, Mori L, Giacche M, et al. Genetic mutation screening in an Italian cohort of nonsyndromic pheochromocytoma/paraganglioma patients. Ann N Y Acad Sci. 2006;1073:156–65.
Baysal BE, Willett-Brozick JE, Lawrence EC, et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet. 2002;39(3):178–83.
• Eisenhofer G, Pacak K, Huynh TT, et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer. 2011;18(1):97–111. This article establishes mutation-dependent phenotypic differences in catecholamine biosynthetic and secretory pathways in different hereditary forms of pheochromocytoma. These differences parallel differences indicated by gene expression profiling studies.
Eisenhofer G, Bornstein SR, Brouwers FM, et al. Malignant pheochromocytoma: current status and initiatives for future progress. Endocr Relat Canc. 2004;11(3):423–36.
•• Eisenhofer G, Lenders JW, Siegert G, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2011 Oct 28 (Epub ahead of print). In this report, plasma concentrations of methoxytyramine are reported as a novel biomarker of malignant pheochromocytoma. The study also establishes the relative contributions of tumor size and extra-adrenal tumor location as risk factors for metastatic disease and the contributions of both to the high risk of malignancy associated with SDHB mutations.
Remine W, Chong G, van Heerden J, et al. Current management of pheochromocytoma. Ann Surg. 1974;179:740–8.
John H, Ziegler WH, Hauri D, Jaeger P. Pheochromocytomas: can malignant potential be predicted? Urology. 1999;53(4):679–83.
Shen WT, Sturgeon C, Clark OH, et al. Should pheochromocytoma size influence surgical approach? a comparison of 90 malignant and 60 benign pheochromocytomas. Surgery. 2004;136(6):1127–9.
Ayala-Ramirez M, Feng L, Johnson MM, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011;96(3):717–25.
Feng F, Zhu Y, Wang X, et al. Predictive factors for malignant pheochromocytoma: analysis of 136 patients. J Urol. 2011;185(5):1583–90.
Park J, Song C, Park M, et al. Predictive characteristics of malignant pheochromocytoma. Korean J Urol. 2011;52(4):241–6.
Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63(17):5615–21.
Brouwers FM, Eisenhofer G, Tao JJ, et al. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91(11):4505–9.
Tippett PA, McEwan AJ, Ackery DM. A re-evaluation of dopamine excretion in phaeochromocytoma. Clin Endocrinol (Oxf). 1986;25(4):401–10.
Januszewicz W, Wocial B, Januszewicz A, et al. Dopamine and dopa urinary excretion in patients with pheochromocytoma–diagnostic implications. Blood Pres. 2001;10(4):212–6.
van der Harst E, de Herder WW, de Krijger RR, et al. The value of plasma markers for the clinical behaviour of phaeochromocytomas. Eur J Endocrinol. 2002;147(1):85–94.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eisenhofer, G. Screening for Pheochromocytomas and Paragangliomas. Curr Hypertens Rep 14, 130–137 (2012). https://doi.org/10.1007/s11906-012-0246-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-012-0246-y
Keywords
- Pheochromocytoma
- Paraganglioma
- Screening
- Incidentaloma
- Adrenal
- Extra-adrenal
- Metastases
- Malignant
- Chromaffin cell tumor
- Diagnosis
- Normetanephrine
- Metanephrine
- Methoxytyramine
- Norepinephrine
- Epinephrine
- Dopamine
- Catecholamines
- von Hippel-Lindau
- Multiple endocrine neoplasia type 2
- Neurofibromatosis
- Succinate dehydrogenase