
Abstract
Apolipoprotein L1 (APOL1) gene association studies and results of the African American Study of Kidney Disease and Hypertension are disproving the longstanding concept that mild to moderate essential hypertension contributes substantially to end-stage renal disease susceptibility in African Americans. APOL1 coding variants underlie a spectrum of kidney diseases, including that attributed to hypertension (labeled arteriolar or hypertensive nephrosclerosis), focal segmental glomerulosclerosis, and HIV-associated nephropathy. APOL1 nephropathy risk variants persist because of protection afforded from the parasite that causes African sleeping sickness. This breakthrough will lead to novel treatments for hypertensive African Americans with low-level proteinuria, for whom effective therapies are lacking. Furthermore, APOL1 nephropathy risk variants contribute to racially variable allograft survival rates after kidney transplantation and assist in detecting nondiabetic forms of nephropathy in African Americans with diabetes. Discovery of APOL1-associated nephropathy was a major success of the genetics revolution, demonstrating that secondary hypertension is typically present in nondiabetic African Americans with nephropathy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
End-stage renal disease (ESRD) occurs four times more often in African Americans than in European Americans [1], with life-time risk approximating 7.5% in African Americans and only 2% in European Americans [2]. Nondiabetic chronic kidney disease (CKD), particularly hypertension-attributed nephropathy, focal segmental glomerulosclerosis (FSGS), and HIV-associated collapsing glomerulopathy (also called HIV-associated nephropathy or HIVAN) demonstrates pronounced racial differences in incidence, with up to 50-fold higher rates of HIVAN in African Americans than in European Americans [1].
Until recently, the cause of the higher nephropathy risk in individuals of African ancestry remained elusive. Many investigators proposed environmental risk factors as major contributors, with particular emphasis on generally lower socioeconomic status, lack of access to adequate healthcare, and higher blood pressures in African Americans [3, 4]. Modern molecular genetic techniques have proven that variation in the apolipoprotein L1 gene (APOL1) explains the excess risk of nondiabetic ESRD in African Americans [5••, 6•, 7]. This breakthrough is leading to the reclassification of FSGS and other nondiabetic forms of nephropathy in African Americans, including forms long attributed to high blood pressure [8].
The Link between Hypertension and Nephropathy
American nephrologists classify nearly 35% of cases of ESRD in African Americans as being due to essential hypertension, so-called hypertensive nephrosclerosis or arteriolar nephrosclerosis [1]. Diagnoses in the US Renal Data System (USRDS) registry are provided by nearly 6,000 practicing clinical nephrologists without standardized diagnostic criteria and often without biopsy material. These diagnoses are frequently incorrect [9]. Accelerated and malignant forms of hypertension clearly initiate kidney disease, but direct evidence linking mild to moderate hypertension with the initiation of nephropathy in African Americans is weak [10]. Hypertension-attributed nephropathy is typically diagnosed in nondiabetic African Americans with unknown causes of kidney disease; renal histology is usually lacking, and diagnoses are often made without regard to the level of proteinuria [11].
The controversy has been fueled by the frequent absence of clinical data in African Americans with hypertension-attributed kidney disease [12]. A carefully performed study demonstrated marked physician bias, whereby renal diagnoses were often based on the patient’s race rather than objective clinical and laboratory data [13]. Results of population-based epidemiologic studies involving predominantly hypertensive European Americans have frequently been extrapolated to the dialysis clinic, where they appear to be less applicable [14]. The possibility that high blood pressure in CKD patients was secondary to undiagnosed primary kidney diseases, as now appears to be a common scenario, typically was not considered [15].
Firmer evidence that essential hypertension did not commonly initiate nephropathy in African Americans came from renal histologic studies revealing the lack of correlation between arteriolar changes and systemic blood pressure in participants in the African American Study of Kidney Disease (AASK) Trial [16]. These individuals were clinically diagnosed with “hypertensive nephropathy.” Kidney biopsies in a subset of 39 AASK participants revealed segmental or focal global glomerulosclerosis (FGGS), interstitial fibrosis, and arteriosclerosis/arteriolosclerosis [16]. Racially variable patterns of glomerulosclerosis have also been reported. Solidified glomerulosclerosis is present in African Americans more often than in European Americans [17]. Finally, the failure of blood pressure control to halt nephropathy progression in hypertensive African Americans, despite evidence of benefit in European Americans, strongly suggested that the underlying causes of nondiabetic nephropathy attributed to hypertension differed between the races [18, 19].
Excluding polycystic kidney disease, hereditary nephritis, and urologic disease, more than 30% of African Americans initiating renal replacement therapy who have common forms of nephropathy (e.g., clinically diagnosed hypertensive ESRD, diabetic ESRD, and chronic glomerular disease) have close relatives on dialysis, and many more have relatives with silent kidney disease manifesting as proteinuria and/or reduced glomerular filtration rate (GFR) [20, 21]. The pronounced familial aggregation of advanced kidney disease in African Americans, far stronger than that in European Americans [22], provided a major clue to the existence of an overarching nephropathy susceptibility gene [10, 23]. Most striking was the clustering of disparate forms of kidney disease within single African American families, including ESRD attributed to hypertension, primary chronic glomerular diseases (FSGS), HIVAN, systemic lupus erythematosus (SLE), and diabetes mellitus [24, 25]. The existence of a major “nephropathy susceptibility gene” underlying several common forms of kidney disease in African Americans was first proposed in the early 1990s [24, 26], but the concept was widely rebuked [27].
Hypertension: The Chicken or the Egg in Arteriolar Nephrosclerosis?
The debate over whether hypertension or nephropathy developed first in the vicious cycle of hypertension-attributed kidney disease contributed to the performance of the AASK Trial, sponsored by the National Institutes of Health [28]. AASK tested the benefits of drugs in three antihypertensive classes—angiotensin-converting enzyme (ACE) inhibitors (ramipril), calcium channel blockers (amlodipine), and beta-blockers (metoprolol)—as well as two levels of blood pressure control—low (mean arterial blood pressure goal <92 mm Hg) versus usual (102–107 mm Hg)—for nephropathy progression rates in 1,094 nondiabetic, hypertensive African Americans who met criteria for putative “hypertensive nephrosclerosis.” Disease criteria included an initial estimated GFR of 20 to 65 mL/min/1.73 m2 and proteinuria less than 2.5 g/day [28].
After 5 years, the AASK Trial demonstrated that baseline proteinuria was associated with more rapid progression of nephropathy, but the lower blood pressure target failed to slow nephropathy progression more than the usual target [29]. After completion of the AASK Trial, the remaining participants who had not reached a primary study end point were followed in the AASK Cohort phase; all used ACE inhibition with ramipril and the lower blood pressure goal. The AASK Cohort study subsequently confirmed the lack of benefit of the lower blood pressure goal and ACE inhibition in the overall study sample. Nearly 60% of participants reached a primary study end point (death, dialysis, or doubling of serum creatinine concentration) after 10 years [30••]. These interventions appeared to be somewhat beneficial in slowing nephropathy progression rates among participants with baseline proteinuria, just as in patients with proteinuric glomerular diseases including FSGS [31]. However, it is now clear that lower blood pressures and ACE inhibition are ineffective in slowing nephropathy progression in hypertensive African Americans without proteinuria. We believe the results of this important study sever the link between high blood pressure and nephropathy in African Americans [32].
Chromosome 22q13.1 Genetic Associations in Nondiabetic Nephropathy
The AASK was initiated to identify treatment options for the chronic kidney disease that was attributed to hypertension. As such, AASK inclusion criteria are useful to capture a standard, accepted phenotype for hypertension-attributed nephropathy. Recently, APOL1 and MYH9 nephropathy risk variants were proven to be strongly associated with kidney disease in AASK participants, linking this disorder with FSGS, hypertension-attributed ESRD, and HIVAN [33]. Mapping by admixture linkage disequilibrium (MALD), also called admixture mapping, has proven to be useful for detecting genetic variants contributing to disease risk in admixed populations, in which ancestral populations have different disease frequencies [34, 35]. African Americans are an admixed population with approximately 82% African and 18% European ancestry, and kidney disease is far more common in individuals with African ancestry.
Kopp et al. [36] and Kao et al. [37] first applied MALD to kidney disease and detected an impressive genetic association between the non-muscle myosin heavy chain 9 gene (MYH9) E1 (Extended-1) haplotype and idiopathic FSGS, nondiabetic ESRD, and HIV-associated collapsing glomerulopathy in African Americans. The observation was soon extended to patients with C1q-associated collapsing glomerulosclerosis [38], clinically diagnosed hypertension-attributed ESRD [39], and type 2 diabetes–attributed ESRD [40]. In retrospect, the association with diabetic nephropathy likely resulted from inclusion of diabetic subjects with nondiabetic (proteinuric) nephropathy in the FSGS spectrum of disease [41, 42•]. MYH9 risk alleles were not associated with essential hypertension per se in African Americans who lacked advanced nephropathy [43].
Within 2 years of the MYH9 discovery, Genovese, Friedman, and colleagues demonstrated that two coding variants in APOL1 (termed G1: non-synonymous coding variant 342 G:384 M and G2: a 6 bp deletion) had stronger genetic association with nondiabetic nephropathy than the MYH9 E1 haplotype [5••]. At this time, coding variants in MYH9 had not been detected despite extensive fine mapping efforts [44]. Newly available single nucleotide polymorphisms (SNPs) in Yoruba from the “1,000 Genomes” Project led to detection of APOL1 association. Strong linkage disequilibrium between the SNPs in these adjacent genes on chromosome 22q13.1 contributed to the initial difficulty in detecting the causative gene; strong selective pressures were also felt to be operative in this region [5••, 45•]. The APOL1 association has been independently replicated [6•]. The ApoL1 protein is associated with relative immunity to trypanosomal infection and protection from African sleeping sickness [46, 47]. Nephropathy risk variants were shown to encode proteins that effectively lysed Trypanosoma brucei rhodesiense, a property absent in those with wild-type (non-nephropathy) variants [5••]. Centuries ago, human biologic and genome evolution led to positive selection for APOL1 nephropathy risk variants. The molecular basis of this adaptive evolution was from the protection conferred by these variants against the deadly parasitic disease transmitted by the tsetse fly in sub-Saharan Africa.
Idiopathic FSGS is strongly associated with APOL1 G1 and G2 variants in an autosomal recessive fashion [5••, 7]. This finding is among the most impressive genetic associations in complex human disease. Individuals inheriting two APOL1 nephropathy risk variants (G1 + G1, G2 + G2, or G1 + G2) have a 10.5-fold increased risk for FSGS. Clinically diagnosed hypertension-attributed ESRD is also strongly associated (odds ratio 7.3 recessive; P = 10−63). Together, hypertension-attributed ESRD and various forms of FSGS account for 40% of incident ESRD cases in African Americans [1]. As stated, APOL1 risk alleles were also strongly associated with kidney disease in AASK participants, demonstrating that the kidney disease in AASK resides in the FSGS–FGGS spectrum of APOL1-associated nephropathy and is unlikely to relate to renal damage from high blood pressure.
Whether MYH9 plays a residual role in renal disease susceptibility is under active investigation. Genovese et al. performed a conservative statistical analysis and concluded that essentially all of the chromosome 22q nephropathy risk resided in APOL1 [5••]. Other investigators continue to detect an MYH9 effect in their study samples after adjustment for APOL1 G1 and G2 risk variants (personal communication 2011: JB Kopp – NIH FSGS; CD Langefeld – Wake Forest School of Medicine; WH Kao – Family Investigation of Nephropathy and Diabetes [FIND]). APOL1 G1 and G2 nephropathy risk variants are exceedingly rare in European Americans, whereas MYH9 variants appear to contribute to nephropathy risk in several European-derived populations [48, 49•, 50]. Because of strong selective forces and linkage disequilibrium in this region, it remains possible that MYH9 association relates to SNPs in nearby genes (perhaps other APOL1 coding variants or members of the APOL2-6 gene family). Deep sequencing of the APOL-MYH9 gene region will be necessary to clarify this question. Based on the weakly dominant APOL1 inheritance that was observed in hypertension-attributed ESRD, we feel it is likely that additional undetected nephropathy risk variants reside in the region [5••]. In this fashion, G1 or G2 would appear to have a dominant effect, but a second (undetected) risk variant would be present and maintain recessive inheritance. These remaining variants are likely to be less frequent than APOL1 G1 and G2.
Clinical Applications of APOL1 Genotyping in Renal Transplantation
Beyond the exciting possibility of identifying the cause, and ultimately a cure, for the kidney disease that has historically been attributed to hypertension in African Americans, APOL1 genotyping is proving to have important clinical applications in the arena of transplant nephrology.
Race-driven outcomes exist in renal transplantation [51]. Kidneys donated by African Americans function for significantly shorter time periods than kidneys donated by European Americans [52–55]. African American live kidney donors are also more likely than other kidney donors to develop severe nephropathy after donation [56].
We reported that the risk of early graft failure in African American donated kidneys relates predominantly to APOL1 genetic variation: deceased-donor kidneys harboring two APOL1 risk variants function for significantly shorter periods than those with less than two risk variants [57••]. The effect of APOL1 on renal allograft survival is far stronger than the effects observed for HLA matching, panel reactive antibodies (PRA), standard (vs expanded)-criteria donation, and cold ischemia time. Evaluation of ancestry informative markers across the genome localized the risk to APOL1, confirming that donor race per se is not the cause. In fact, African American donor kidneys with less than two APOL1 risk variants functioned for a duration similar to that of kidneys from European American donors. Among failing kidneys from donors with two APOL1 risk variants, 75% had histologic lesions compatible with APOL1-associated nephropathy, compared with 11.8% of failing allografts from non-risk donors.
We believe that APOL1 risk variants likely contribute to the more frequent development of ESRD in living African American donors, relative to European Americans [57••]. Although not yet proven, 50% reductions in nephron mass in the presence of two APOL1 risk variants is likely to accelerate the course of renal injury in at-risk individuals. Screening donor kidneys for APOL1 risk variants has the potential to improve long-term renal allograft survival and simultaneously reduce the rates of subsequent ESRD in African American living kidney donors [58, 59].
Kidney Disease Screening
The usefulness of screening for APOL1 risk variants in the general African American population remains unknown [60]. Of African American chromosomes (not individuals), 30% contain an APOL1 nephropathy risk variant and 70% do not [5••, 7]. As such, 49% (70% × 70%) of African Americans lack G1 and G2 variants; 51% inherit one or two risk variants, and approximately 12% of African Americans have two risk variants and are at high risk for kidney disease. As the majority of African Americans develop high blood pressure by late adulthood and APOL1 (and MYH9) risk variants do not appear to be associated with hypertension, there is no a priori reason to screen only hypertensive individuals [43]. Until effective treatments for APOL1-associated nephropathy are available, general population screening would seem to have little benefit except in research settings, genetic studies in ESRD patients who lack definitive diagnoses based on renal biopsy material (see below), and potentially for screening African American kidney donors.
Not every individual who inherits two APOL1 risk variants will develop ESRD, so a search for “second hits” (either gene-gene or gene-environment interactions) has begun [61]. HIV infection is an important environmental factor that can lead to the development of APOL1-associated nephropathy, and highly active anti-retroviral therapy (HAART) appears to provide protection. Preventing (or treating) environmental factors or exposures that may interact with risk genes has the potential to inhibit development (or slow the progression) of nondiabetic etiologies of nephropathy in individuals at risk based on APOL1.
APOL1 and MYH9 genotyping provides the opportunity to detect African Americans with ESRD who have a high likelihood of renal lesions in the spectrum of FSGS and FGGS. This is particularly important in patients with systemic disorders such as type 2 diabetes mellitus and SLE, both common in African Americans, in whom proteinuric kidney disease can be related to the systemic process (diabetic nephropathy or lupus nephritis), idiopathic FSGS, or a combination of both lesions. We reported that a diabetic patient with two chromosome 22q risk variants had FSGS on kidney biopsy, despite a clinical diagnosis of diabetic nephropathy [41]. This finding has relevance for clinical trials treating nephropathy associated with diabetes or lupus: African Americans with two APOL1 risk variants may need to be excluded or evaluated separately to ensure that idiopathic FSGS does not impact study results [62].
In a similar fashion, we demonstrated the utility of stratifying African Americans with type 2 diabetes mellitus and ESRD based on the presence of chromosome 22q risk variants in order to detect diabetic nephropathy susceptibility genes [42•]. A prior genome-wide association study (GWAS) for type 2 diabetic nephropathy in African Americans failed to detect evidence of association of the 4.1 protein ezrin, radixin, moesin (FERM) domain containing 3 locus gene (FRMD3) [63], which was previously associated with type 1 diabetic nephropathy in European Americans [64]. Interactions appeared to exist between MYH9 and APOL1 risk variants and FRMD3 in a genome-wide gene-gene interaction analysis [42•]. Opposing effects of FRMD3 SNPs were observed in chromosome 22q risk homozygotes and non-risk homozygotes, suggesting the presence of different causes of kidney disease between these groups. In chromosome 22q non-risk homozygotes, likely enriched for true diabetic nephropathy, significant association was observed with multiple FRMD3 SNPs. However, in MYH9 and APOL1 risk homozygotes, enriched for nondiabetic nephropathy (FSGS), evidence of FRMD3 association was not detected. We propose that genotyping APOL1 nephropathy risk variants can assist in the genetic dissection of FSGS-related disorders in African Americans with systemic diseases that may cause proteinuria and kidney failure.
Potential Mechanisms of APOL1-Associated Nephropathy
APOL1 nephropathy risk variants are trypanolytic via the effect of the ApoL1 protein, which functions as a chloride channel [47]. Once taken up by the trypanosome, ApoL1 undergoes a conformational change and inserts into the lysosomal membrane. Chloride permeability increases, with resultant swelling and lysosomal rupture and death of the parasite. However, the mechanism whereby APOL1 gene expression in renal cells or ApoL1 proteins in the circulation (either free or bound to high-density lipoprotein [HDL] cholesterol) contributes to nephropathy risk remains unknown.
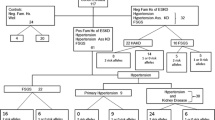
APOL1 encodes ApoL1, a secretory protein that associates with HDL cholesterol in the plasma [65]. Three mechanisms can be proposed by which ApoL1 variants contribute, either singly or in combination, to the development of nondiabetic nephropathy (Fig. 1). Altered plasma HDL particle concentrations appear to result from homozygosity for APOL1 risk variants. These changes could lead to kidney disease via damage to the renal microcirculation. We recently measured HDL subclass concentrations in 73 African American first-degree relatives of patients with nondiabetic ESRD to detect differences that potentially may contribute to development of kidney disease [66]. HDL subclass concentrations were measured using nuclear magnetic resonance spectroscopy, and additive effects of the number of APOL1 risk variants on natural logarithm transformed HDL subclass concentrations were computed. Mean ± SD medium-sized HDL concentrations were significantly lower for each additional APOL1 risk variant: 9.0 ± 5.6 μmol/L with two risk variants; 10.1 ± 5.5 μmol/L with one risk variant; 13.1 ± 8.2 μmol/L with no risk variants (P = 0.0192 unadjusted, P = 0.0190 triglyceride-adjusted). Hence, lower medium-sized HDL subclass concentrations were present with increasing numbers of APOL1 nephropathy risk variants. Potential mechanistic roles for altered medium-sized HDL particle concentrations on APOL1-associated renal microvascular disease remain to be determined. Similar patterns of HDL particle concentration contribute to coronary artery disease susceptibility, suggesting potential effects on the vascular endothelium [67].

Potential mechanistic pathways elicited by APOL1 variants that may contribute to the development of nondiabetic nephropathy. FGGS focal global glomerulosclerosis; FSGS focal segmental glomerulosclerosis; HDL high-density lipoprotein
A second possibility is that nephropathy-associated ApoL1 variant proteins bind to HDL particles less avidly, circulate freely, cross the glomerular filtration barrier, and lead to glomerulosclerosis and/or interstitial fibrosis. This hypothesis is strengthened by the observation that circulating factors are often responsible for recurrent FSGS after renal transplantation [68]. Plasmapheresis removes these circulating factors and may lead to remission of proteinuria [69]. Moreover, ApoL1 has recently been implicated as a circulating factor that may underlie recurrent FSGS after kidney transplantation [70, 71]. This exciting finding could provide novel treatment options, if circulating variant forms of ApoL1 lead to altered glomerular permeability with progressive kidney failure. A third possibility is that nephropathy-associated APOL1 variants may induce apoptosis or autophagy in podocytes, with resultant FSGS or FGGS. Podocytes express ApoL1 mRNA, and the apoptosis regulating B-cell lymphoma 2 gene (Bcl2) is closely related to APOL1 [72]. Determining how the APOL1 association with nondiabetic ESRD induces nephropathy is crucial to unraveling disease pathogenesis and offers hope for curing ESRD in affected African Americans in whom hypertension treatment alone has failed to significantly slow renal disease progression.
Non–Chromosome 22q Hypertensive Nephropathy Genes
The recent APOL1 breakthrough demonstrates that true hypertension-attributed kidney disease occurs less often in African Americans than previously thought. In contrast, renal small blood vessel disease in European Americans appears to occur in relation to high blood pressure, hyperlipidemia, smoking, and a Western lifestyle. Hypertension treatment slows progression of this form of kidney disease in European Americans [18].
However, APOL1 variants are not associated with all nondiabetic kidney disease in African Americans. Polymorphisms in the chromogranin A (CHGA) and methylenetetrahydrofolate reductase (MTHFR) genes have been shown to be associated with renal functional decline in African Americans with hypertension-attributed nephropathy [73, 74]. Similarly, podocin (NPHS2) mutations causing FSGS are occasionally identified in African Americans with hypertension-attributed ESRD [75]. Although APOL1 variants associate with most cases of nondiabetic ESRD, other genes clearly make contributions.
Conclusions
The past few years have seen dramatic changes in our understanding of the disease process that had historically been labeled hypertensive nephrosclerosis. The majority of these cases in African Americans are associated with coding variants in the APOL1 gene on chromosome 22q. The AASK Trial and Cohort studies revealed the lack of efficacy of aggressive blood pressure reduction using ACE inhibitor-based treatment regimens in nondiabetic, hypertensive African Americans with low-level proteinuria. APOL1 has proven to be associated with kidney disease in AASK participants. Whether the inciting renal lesion in these forms of nephropathy resides in the glomerulus (FSGS or FGGS), the renal interstitium, or small blood vessels is yet to be determined, but it is clear that most cases of hypertension-attributed nephropathy are initiated by primary kidney diseases, and blood pressure is elevated secondarily. Molecular genetic techniques have once and for all resolved the issue of what came first: nephropathy clearly precedes hypertension in the majority of African American patients with nondiabetic nephropathy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
U.S. Renal Data System. USRDS 2010 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disease, Bethesda, MD. 121–132. 2010.
Kiberd BA, Clase CM. Cumulative risk for developing end-stage renal disease in the US population. J Am Soc Nephrol. 2002;13:1635–44.
McClellan WM, Newsome BB, McClure LA, et al. Poverty and racial disparities in kidney disease: the REGARDS study. Am J Nephrol. 2010;32:38–46.
Evans K, Coresh J, Bash LD, et al. Race differences in access to health care and disparities in incident chronic kidney disease in the US. Nephrol Dial Transplant. 2011;26:899–908.
•• Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010, 329:841–845. This landmark report revealed that two coding variants in the apolipoprotein L1 gene (APOL1), termed G1 and G2, were strongly associated with FSGS and nondiabetic forms of ESRD. These diseases were previously attributed to variation in the adjacent non-muscle myosin heavy chain 9 gene (MYH9). The protein product of nephropathy-associated APOL1 variants lysed Trypanosoma brucei rhodesiense, a cause of African sleeping sickness.
• Tzur S, Rosset S, Shemer R, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 2010, 128:345–350. This report also revealed that variation in the APOL1 gene contributed to nondiabetic ESRD susceptibility in African Americans. The G1 nephropathy risk variant was detected.
Freedman BI, Kopp JB, Langefeld CD, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. 2010;21:1422–6.
Howie AJ. Problems with ‘focal segmental glomerulosclerosis’. Pediatr Nephrol. 2011;26:1197–205.
Zarif L, Covic A, Iyengar S, et al. Inaccuracy of clinical phenotyping parameters for hypertensive nephrosclerosis. Nephrol Dial Transplant. 2000;15:1801–7.
Freedman BI, Iskandar SS, Appel RG. The link between hypertension and nephrosclerosis. Am J Kidney Dis. 1995;25:207–21.
Freedman BI, Sedor JR. Hypertension-associated kidney disease: perhaps no more. J Am Soc Nephrol. 2008;19:2047–51.
Schlessinger SD, Tankersley MR, Curtis JJ. Clinical documentation of end-stage renal disease due to hypertension. Am J Kidney Dis. 1994;23:655–60.
Perneger TV, Whelton PK, Klag MJ, Rossiter KA. Diagnosis of hypertensive end-stage renal disease: effect of patient’s race. Am J Epidemiol. 1995;141:10–5.
Klag MJ, Whelton PK, Randall BL, et al. Blood pressure and end-stage renal disease in men. N Engl J Med. 1996;334:13–8.
Kestenbaum B, Rudser KD, de Boer I. Differences in kidney function and incident hypertension: the multi-ethnic study of atherosclerosis. Ann Intern Med. 2008;148:501–8.
Fogo A, Breyer JA, Smith MC, et al. Accuracy of the diagnosis of hypertensive nephrosclerosis in African Americans: a report from the African American Study of Kidney Disease (AASK) Trial. AASK Pilot Study Investigators. Kidney Int. 1997;51:244–52.
Marcantoni C, Ma LJ, Federspiel C, Fogo AB. Hypertensive nephrosclerosis in African Americans versus Caucasians. Kidney Int. 2002;62:172–80.
Shulman NB, Ford CE, Hall WD, et al. Prognostic value of serum creatinine and effect of treatment of hypertension on renal function. Results from the hypertension detection and follow-up program. The Hypertension Detection and Follow-up Program Cooperative Group. Hypertension. 1989;13:I80–93.
Walker WG, Neaton JD, Cutler JA, et al. Renal function change in hypertensive members of the Multiple Risk Factor Intervention Trial. Racial and treatment effects. The MRFIT Research Group. JAMA. 1992;268:3085–91.
Freedman BI, Soucie JM, McClellan WM. Family history of end-stage renal disease among incident dialysis patients. J Am Soc Nephrol. 1997;8:1942–5.
Freedman BI, Volkova NV, Satko SG, et al. Population-based screening for family history of end-stage renal disease among incident dialysis patients. Am J Nephrol. 2005;25:529–35.
Spray BJ, Atassi NG, Tuttle AB, Freedman BI. Familial risk, age at onset, and cause of end-stage renal disease in white Americans. J Am Soc Nephrol. 1995;5(10):1806–10.
Freedman BI. Susceptibility genes for hypertension and renal failure. J Am Soc Nephrol. 2003;14(7 Suppl 2):S192–4.
Freedman BI, Spray BJ, Tuttle AB, Buckalew Jr VM. The familial risk of end-stage renal disease in African Americans. Am J Kidney Dis. 1993;21:387–93.
Freedman BI. End-stage renal failure in African Americans: insights in kidney disease susceptibility. Nephrol Dial Transplant. 2002;17(2):198–200.
Freedman BI. Renal microvascular susceptibility in African American pedigrees. Transplant Proc. 1993;25:2423–5.
Cooper RS, Kaufman JS, Ward R. Race and genomics. N Engl J Med. 2003;348:1166–70.
Appel LJ, Middleton J, Miller III ER, et al. The rationale and design of the AASK cohort study. J Am Soc Nephrol. 2003;14:S166–72.
Wright Jr JT, Bakris G, Greene T, et al. Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. JAMA. 2002;288:2421–31.
•• Appel LJ, Wright JT Jr, Greene T, et al. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med 2010, 363:918–929. This report contained the final AASK Cohort Study results. Strict blood pressure control using ACE inhibition failed to slow or halt nephropathy progression in African Americans thought to have hypertensive kidney disease and lacking proteinuria. Some benefit was seen in those participants with elevated baseline urinary protein excretion.
Korbet SM. Angiotensin antagonists and steroids in the treatment of focal segmental glomerulosclerosis. Semin Nephrol. 2003;23:219–28.
Freedman BI, Sedor JR. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med. 2010;363:2565–6.
Astor B, Lipkowitz MS, Kao WHL, Parekh R, Choi MH, Kopp JB, et al. MYH9 variations are associated with chronic kidney disease progression in the African American Study of Hypertension and Kidney Diseases [abstract]. J Am Soc Nephrol. 2010;21:81A.
Smith MW, Patterson N, Lautenberger JA, et al. A high-density admixture map for disease gene discovery in African Americans. Am J Hum Genet. 2004;74:1001–13.
Divers J, Moossavi S, Langefeld CD, Freedman BI. Genetic admixture: a tool to identify diabetic nephropathy genes in African Americans. Ethn Dis. 2008;18:384–8.
Kopp JB, Smith MW, Nelson GW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008;40:1175–84.
Kao WH, Klag MJ, Meoni LA, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet. 2008;40:1185–92.
Reeves-Daniel AM, Iskandar SS, Bowden DW, et al. Is collapsing C1q nephropathy another MYH9-associated kidney disease? A case report. Am J Kidney Dis. 2010;55:e21–4.
Freedman BI, Hicks PJ, Bostrom MA, et al. Polymorphisms in the non-muscle myosin heavy chain 9 gene (MYH9) are strongly associated with end-stage renal disease historically attributed to hypertension in African Americans. Kidney Int. 2009;75:736–45.
Freedman BI, Hicks PJ, Bostrom MA, et al. Non-muscle myosin heavy chain 9 gene MYH9 associations in African Americans with clinically diagnosed type 2 diabetes mellitus-associated ESRD. Nephrol Dial Transplant. 2009;11:3366–71.
Gopalakrishnan I, Iskandar SS, Daeihagh P, et al. Coincident idiopathic focal segmental glomerulosclerosis collapsing variant and diabetic nephropathy in an African American homozygous for MYH9 risk variants. Hum Pathol. 2011;42:291–4.
• Freedman BI, Langefeld CD, Lu L, et al. Differential effects of MYH9 and APOL1 risk variants on FRMD3 association with diabetic ESRD in African Americans. PLoS Genet 2011, 7:e1002150. This gene-gene interaction analysis assessed APOL1/MYH9 SNPs and nearly one million other genetic markers across the genome in African Americans with clinically diagnosed type 2 diabetic ESRD. Those with two MYH9 (or APOL1) risk variants did not demonstrate association with FRMD3, a diabetic nephropathy gene. Markers in FRMD3 were robustly associated in those lacking two chromosome 22q risk variants, revealing the potential to “genetically dissect” nondiabetic from diabetic ESRD in African Americans based on the presence of two MYH9 or APOL1 risk variants.
Freedman BI, Kopp JB, Winkler CA, et al. Polymorphisms in the nonmuscle myosin heavy chain 9 gene (MYH9) are associated with albuminuria in hypertensive African Americans: the HyperGEN study. Am J Nephrol. 2009;29:626–32.
Nelson GW, Freedman BI, Bowden DW, et al. Dense mapping of MYH9 localizes the strongest kidney disease associations to the region of introns 13 to 15. Hum Mol Genet. 2010;19:1805–15.
• Oleksyk TK, Nelson GW, An P, et al. Worldwide distribution of the MYH9 kidney disease susceptibility alleles and haplotypes: evidence of historical selection in Africa. PLoS One 2010, 5:e11474. This study assessed SNPs in the MYH9 E haplotype block in a worldwide population survey of major continental populations using International HapMap Project and the Human Genome Diversity Panel data. The worldwide distribution and frequencies of risk/protective haplotypes were presented, relevant to public health implications, including in regions of the world with a high prevalence of HIV infection.
Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. 2003;422:83–7.
Pays E, Vanhollebeke B. Human innate immunity against African trypanosomes. Curr Opin Immunol. 2009;21:493–8.
Pattaro C, Aulchenko YS, Isaacs A, et al. Genome-wide linkage analysis of serum creatinine in three isolated European populations. Kidney Int. 2009;76:297–306.
• O’Seaghdha CM, Parekh RS, Hwang SJ, et al. The MYH9/APOL1 region and chronic kidney disease in European-Americans. Hum Mol Genet 2011, 20:2450–2456. MYH9 E1 haplotype SNP rs4821480 and 282 SNPs in the MYH9/APOL1 region were tested for association with subclinical kidney disease in 13,133 European-derived participants from the Framingham Heart and Atherosclerosis Risk in Communities Studies. In a meta-analysis, only rs4821480 was associated with nondiabetic kidney disease. APOL1 G1 and G2 risk variants did not account for the observed MYH9 signal, as they were exceedingly rare in these European-derived populations. This suggests that MYH9 SNP rs4821480 is associated with risk of nondiabetic nephropathy in individuals of European ancestry.
Cooke JN, Bostrom MA, Hicks PJ, Ng MC, Comeau ME, Divers J, Langefeld CD, Freedman BI, Bowden DW. Polymorphisms in MYH9 are associated with diabetic nephropathy in European Americans. Nephrol Dial Transplant 2011 Oct 3 (Epub ahead of print). doi: 10.1093/ndt/gfr522.
Steiner RW. ‘Normal for now’ or ‘at future risk’: a double standard for selecting young and older living kidney donors. Am J Transplant. 2010;10:737–41.
Meier-Kriesche HU, Port FK, Ojo AO, et al. Effect of waiting time on renal transplant outcome. Kidney Int. 2000;58:1311–7.
Swanson SJ, Hypolite IO, Agodoa LY, et al. Effect of donor factors on early graft survival in adult cadaveric renal transplantation. Am J Transplant. 2002;2:68–75.
Chakkera HA, O’Hare AM, Johansen KL, et al. Influence of race on kidney transplant outcomes within and outside the Department of Veterans Affairs. J Am Soc Nephrol. 2005;16:269–77.
Callender CO, Cherikh WS, Traverso P, et al. Effect of donor ethnicity on kidney survival in different recipient pairs: an analysis of the OPTN/UNOS database. Transplant Proc. 2009;41:4125–30.
Cherikh WS, Young CJ, Kramer BF, et al. Ethnic and gender related differences in the risk of end-stage renal disease after living kidney donation. Am J Transplant. 2011;11:1650–5.
•• Reeves-Daniel AM, Depalma JA, Bleyer AJ, et al. The APOL1 gene and allograft survival after kidney transplantation. Am J Transplant 2011, 11:1025–1030. Allograft survival was significantly impacted by the presence of APOL1 variants in African American deceased kidney donors. Kidneys donated by African Americans with two APOL1 risk variants functioned for shorter durations than those donated by individuals with fewer than two risk variants, and African American donor kidneys without two APOL1 risk variants functioned as long as those donated by European Americans. Hence, donor APOL1 genotype, not donor race, explained racial differences in renal allograft survival. If replicated, this study may alter practice patterns, including selection of (a) African American live kidney donors at lower risk for subsequent nephropathy, and (b) deceased kidneys with higher likelihood for long-term function.
Davis CL, Cooper M. The state of U.S. living kidney donors. Clin J Am Soc Nephrol. 2010;5:1873–80.
Srinivas TR. Defining living kidney donor ESRD risk-looking beyond race and gender. Am J Transplant. 2011;11:1556–8.
Kopp JB, Winkler CA, Nelson GW. MYH9 genetic variants associated with glomerular disease: What is the role for genetic testing? Semin Nephrol. 2010;30:409–17.
Bostrom MA, Freedman BI. The spectrum of MYH9-associated nephropathy. Clin J Am Soc Nephrol. 2010;5:1107–13.
Freedman BI, Edberg JC, Comeau ME, et al. The non-muscle myosin heavy chain 9 gene (MYH9) is not associated with lupus nephritis in African Americans. Am J Nephrol. 2010;32:66–72.
McDonough CW, Palmer ND, Hicks PJ, et al. A genome-wide association study for diabetic nephropathy genes in African Americans. Kidney Int. 2011;79:563–72.
Pezzolesi MG, Poznik GD, Mychaleckyj JC, et al. Genome-wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes. 2009;58(6):1403–10.
Gordon SM, Hofmann S, Askew DS, Davidson WS. High density lipoprotein: It’s not just about lipid transport anymore. Trends Endocrinol Metab. 2011;22:9–15.
Freedman BI, Langefeld CD, Murea M, Ma L, Otvos J, Turner JJ, Antinozzi PA, Rocco M, Parks JS. Apolipoprotein L1 nephropathy risk variants associate with HDL subfraction concentration in African Americans. Nephrol Dial Transplant 2011 Sep 19. (Epub ahead of print). doi: 10.1093/ndt/gfr542.
Kuller LH, Grandits G, Cohen JD, et al. Lipoprotein particles, insulin, adiponectin, C-reactive protein and risk of coronary heart disease among men with metabolic syndrome. Atherosclerosis. 2007;195:122–8.
Sharma M, Sharma R, McCarthy ET, Savin VJ. “The FSGS factor:” enrichment and in vivo effect of activity from focal segmental glomerulosclerosis plasma. J Am Soc Nephrol. 1999;10:552–61.
Gohh RY, Yango AF, Morrissey PE, et al. Preemptive plasmapheresis and recurrence of FSGS in high-risk renal transplant recipients. Am J Transplant. 2005;5:2907–12.
Candiano G, Musante L, Zennaro C, et al. Inhibition of renal permeability towards albumin: a new function of apolipoproteins with possible pathogenetic relevance in focal glomerulosclerosis. Electrophoresis. 2001;22:1819–25.
Candiano G, Musante L, Carraro M, et al. Apolipoproteins prevent glomerular albumin permeability induced in vitro by serum from patients with focal segmental glomerulosclerosis. J Am Soc Nephrol. 2001;12:143–50.
Wan G, Zhaorigetu S, Liu Z, et al. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008;283:21540–9.
Salem RM, Cadman PE, Chen Y, et al. Chromogranin A polymorphisms are associated with hypertensive renal disease. J Am Soc Nephrol. 2008;19:600–14.
Fung MM, Salem RM, Lipkowitz MS, et al. Methylenetetrahydrofolate reductase (MTHFR) polymorphism A1298C (Glu429Ala) predicts decline in renal function over time in the African-American Study of Kidney Disease and Hypertension (AASK) Trial and Veterans Affairs Hypertension Cohort (VAHC). Nephrol Dial Transplant 2011 May 25 (Epub ahead of print). PMID: 21613384.
Dusel JA, Burdon KP, Hicks PJ, et al. Identification of podocin (NPHS2) gene mutations in African Americans with nondiabetic end-stage renal disease. Kidney Int. 2005;68:256–62.
Acknowledgments
This work was supported by NIH grants R01 HL56266, RO1 DK070941, and RO1 DK084149.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Freedman, B.I., Murea, M. Target Organ Damage in African American Hypertension: Role of APOL1 . Curr Hypertens Rep 14, 21–28 (2012). https://doi.org/10.1007/s11906-011-0237-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-011-0237-4
Keywords
- African American
- African sleeping sickness
- Arteriolar nephrosclerosis
- APOL1
- Chronic kidney disease
- Dialysis
- End-stage renal disease
- ESRD: Focal segmental glomerulosclerosis
- Genetics
- Glomerulosclerosis
- Hypertension
- Hypertensive nephrosclerosis
- Kidney disease
- Kidney donors
- MYH9
- Nondiabetic nephropathy
- Racial differences
- Trypanosoma brucei rhodesiense
- Transplantation