
Abstract
The renal pressure-natriuresis mechanism is the dominant controller of body fluid balance and long-term arterial pressure. In recent years, it has become clear that the balance of reactive oxygen and nitrogen species within the renal medullary region is a key determinant of the set point of the renal pressure-natriuresis curve. The development of renal medullary oxidative stress causes dysfunction of the pressure-natriuresis mechanism and contributes to the development of hypertension in numerous disease models. The purpose of this review is to point out the known mechanisms within the renal medulla through which reactive oxygen and nitrogen species modulate the pressure-natriuresis response and to update the reader on recent advances in this field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Arthur Guyton [1] was the first to recognize the dominant role of the renal pressure-natriuresis mechanism in the long-term control of arterial pressure. Pressure-natriuresis and pressure-diuresis refer to the ability of the kidney to alter the output of sodium and water respectively in response to alterations in renal arterial pressure. Although multiple physiological mechanisms act in response to altered body fluid balance and changes in arterial blood pressure to modulate cardiac output and total peripheral resistance, normally only a change in sodium and water excretion by the kidneys can bring fluid balance and arterial pressure back to normal levels [1]. In this respect, the renal pressure-natriuresis/diuresis system is considered an infinite-gain system and is the dominant long-term controller of arterial pressure [1].
In salt-resistant individuals, blood pressure rises only minimally in response to a large increase in sodium intake. In such individuals, chronic hypertension can develop only after a change in the set-point of the renal pressure-natriuresis mechanism to higher renal arterial perfusion pressures. Fundamentally, such a change can occur in three ways: 1) changes in glomerular filtration rate, 2) changes in renal blood flow, or 3) changes in tubular reabsorptive activity. Changes in tubular reabsorptive activity underlie the acute natriuretic response to increased arterial pressure observed in experimental settings where renal blood flow and glomerular filtration rate are well autoregulated. Work in our own lab and in others has demonstrated the importance of the renal medullary region in mediating the renal pressure-natriuresis response [2]. It has also become clear that renal medullary levels of reactive oxygen and nitrogen species are capable of modulating the tubular reabsorptive response to increases in arterial pressure and that the levels of outer-medullary reactive oxygen and nitrogen species are unbalanced in many hypertensive disease states and in the salt-sensitive kidney [3]. The purpose of this review is to point out the known mechanisms within the renal medulla through which reactive oxygen and nitrogen species modulate the pressure-natriuresis response and to update the reader on recent advances in this field.
The Renal Medulla and Pressure-Natriuresis
The mechanisms through which renal pressure-natriuresis occurs have become increasingly clear over the past several decades, but certain aspects remain unclear and controversial. Of chief discussion is the physiological mechanism through which arterial pressure is transmitted to the kidney, initiating reduced reabsorption of sodium and water by renal tubules [4]. Data from our own laboratory and others indicate that renal medullary blood flow, unlike whole kidney blood flow and glomerular filtration rate, is relatively poorly autoregulated [5]. Exactly how this pressure and blood flow are delivered to the renal medulla requires clarification, yet it is clear that when renal perfusion pressure is increased, medullary blood flow is also increased, especially in the volume-expanded state [5]. This so-called lack of autoregulation increases vasa recta capillary hydrostatic pressure, which then results in a secondary increase in renal interstitial hydrostatic pressure [6]. The elevated renal interstitial hydrostatic pressure rapidly inhibits sodium reabsorption by the proximal tubules. The reduction in sodium reabsorption in the proximal tubules appears to be mediated by an increase in 20-hydroxyeicosatetraenoic acid (20-HETE), which inhibits Na+-K+-ATPase activity and causes internalization of apical Na+/H+ exchangers [7, 8]. Reductions in sodium reabsorption in deeper nephron segments also occur and are likely to be a result of washout of the medullary solute gradient and reduced passive reabsorption of sodium [9].
An alternative hypothesis suggests that pressure-mediated increases in vascular shear stress stimulate vascular nitric oxide (NO) production and that NO acts as the signal to reduce tubular sodium reabsorption [10]. However, pressure-natriuresis occurs even in the absence of NO production, although it is offset to a higher set point [11]. It is the authors’ view that both NO and superoxide (O −2 ) are important modulators of pressure-natriuresis but are not mediators of this response. It is universally agreed that reactive oxygen and nitrogen species are key modulators of the pressure-natriuresis response, particularly within the renal medullary region.
Nitric Oxide
Mattson et al. [12] were the first to demonstrate that selective inhibition of NO synthase within the renal medulla with the L-arginine analogue N-nitro-L-arginine methyl ester (L-NAME) reduced medullary blood flow and sodium excretion. Chronically administered, this stimulus results in sustained hypertension [13], indicating that endogenous renal medullary NO production is required to prevent the development of hypertension. Supporting this view, intrinsic renal medullary NO levels have been found to be lower in hypertensive rat strains such as the Dahl salt-sensitive rat than in normotensive rat strains [14]. The physiological mechanisms through which renal medullary NO protects from the development of hypertension are likely to vary, but it is known that inhibition of NO production increases the susceptibility of the renal medullary circulation to vasoconstrictor agents such as angiotensin II and vasopressin [15–17]. In situations of reduced medullary NO, administering vasoconstrictor agents in doses that normally are subpressor and do not affect medullary perfusion does result in reduced medullary perfusion and sustained hypertension [14, 18]. These data suggest that a major role of NO is to prevent medullary ischemia and maintain the set point of the renal pressure-natriuresis curve at normotensive levels.
Fenoy et al. [19] first demonstrated that NO blockade blunted increases in renal medullary blood flow and sodium excretion in response to acute increases in renal arterial pressure. In a recent study, Jin et al. [20••] examined both outer medullary and urinary NO2/NO3 (NOx) levels (as an index of NO production) in Sprague-Dawley rats in response to acute increases in renal arterial pressure. In their study, Jin et al. [20••] found that NOx measured in the renal outer medullary region using acute microdialysis techniques rose in response to increased renal arterial pressure, and that these changes in outer medullary NOx were mirrored by parallel changes in urinary NOx excretion (Fig. 1). These data clearly demonstrate that increases in arterial pressure stimulate medullary production of NO. Importantly, decapsulation of the experimental kidney, which reduced renal interstitial hydrostatic pressure and significantly blunted the renal natriuretic response, had no effect on the response of outer medullary NOx [20••]. As medullary NOx was not affected by altering renal interstitial hydrostatic pressure and the tubular natriuretic response, these data suggest that outer medullary NO may be a primary stimulus, associated directly with the increase in renal perfusion pressure, rather than being a secondary response related to the tubular natriuresis [20••].

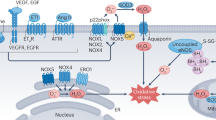
Modulation of pressure-natriuresis by renal medullary reactive oxygen species and nitric oxide (NO). This diagram of the renal medulla illustrates the hypothetical sources of renal medullary reactive oxygen and nitrogen species and their action to stimulate pressure-natriuresis. Increases in renal arterial perfusion pressure result in increased medullary blood flow. Increased medullary blood flow and renal hydrostatic capillary pressure result in increased renal interstitial hydrostatic pressure (RHIP) and capillary shear stress. Increased shear stress on the descending vasa recta epithelial cells stimulates NO production by endothelial NO synthase (NOS). Increased RIHP reduces proximal tubular sodium reabsorption, resulting in increased delivery of sodium and water to the medullary thick ascending limb and stimulating tubular formation of superoxide (O −2 ) and NO. Both tubular and vascular-produced NO stimulate cyclic guanosine 3′5′-monophosphate (cGMP), which acts to further increase RIHP and to inactivate NADPH oxidase, reducing local O −2 production. NO produced by the medullary thick ascending limb or vasa recta endothelium can act on contractile pericytes (blue cell bodies on vasa recta) to reduce vascular resistance and increase medullary blood flow (MBF). As O −2 stimulates sodium transport by the medullary thick ascending limb and inactivates NO, inhibiting O −2 production stimulates tubular natriuresis and increases medullary perfusion. Though cGMP and protein kinase G (PKG) inactivate NADPH oxidase, the net effect of increased renal arterial pressure is an increase in tubular-produced O −2 due to increased tubular flow. Some of this O −2 is converted by superoxide dismutase (SOD) to hydrogen peroxide (H2O2), increasing outer medullary H2O2 levels. The cellular targets of H2O2 are unclear, but H2O2 has an antinatriuretic effect in the renal medulla and may limit the pressure-natriuresis response. Endothelin (Et) produced in response to increases in renal arterial pressure acts on ETB receptors in collecting-duct epithelial cells to stimulate NO production. NO derived from the collecting duct enhances the pressure-natriuresis response, most likely by reducing medullary sodium reabsorption and vascular resistance
NO likely modulates the tubular natriuretic response in a number of ways. It has been shown to inhibit tubular sodium transport in cells of the medullary thick ascending limb, one of the major sodium-transporting epithelia within the renal medullary region [21]. NO may also stimulate renal natriuresis by potentiating the increase in renal interstitial hydrostatic pressure (RIHP), thereby reducing sodium reabsorption in the proximal tubule. In a recent study, Lieb et al. [22•] demonstrate that increases in RIHP stimulate the formation of cyclic guanosine 3′5′-monophosphate (cGMP), which in turn potentiates increases in RIHP and the subsequent natriuretic response (Fig. 1). Therefore, it would appear that NO, via its actions on cGMP formation and medullary perfusion, may stimulate numerous positive feedback loops that act synergistically to increase RIHP and reduce tubular sodium reabsorption. The mechanism underlying the ability of cGMP to increase RIHP remain undetermined. In their publication, Lieb et al. [22•] report that cGMP did not affect renal hemodynamics, suggesting that the increase in RIHP is not related to an increase in medullary perfusion. It should be noted that the technique used by Lieb et al. to measure regional kidney perfusion (laser Doppler flowmetry) differed from those previously used and may not have been able to clearly identify changes in medullary perfusion, so an effect of cGMP to increase medullary perfusion cannot be excluded [4]. By activating multiple synergistic mechanisms, NO may play a key role in modulating the pressure-natriuresis response, enabling rapid and profound decreases in tubular sodium transport in response to very small elevations in arterial pressure.
Nitric oxide synthase (NOS) activity is approximately 25 times greater in the renal medulla than in the renal cortex [23]. The cellular sources of renal medullary NO that contribute to the pressure-natriuresis response are yet to be fully identified. In the study by Jin et al. [20••], in which NOx levels were determined in the outer medulla, the data are consistent with the vasa recta being the source of the NO, as NOx levels mirrored increases in renal medullary blood flow but not the natriuretic response. As increased luminal flow and pressure have been demonstrated to increase descending vasa recta NO production in vitro [24], Jin et al. [20••] speculate that such an increase in outer medullary NO levels could occur as a result of increased blood flow and vascular shear stress in the vasa recta capillaries.
Enzymatically, the greatest source of NO within the renal medullary region is the inner medullary collecting duct [23]. NOS activity in microdissected medullary nephron segments is greatest in the inner medullary collecting duct, followed by the vasa recta; it is lowest in other tubular segments such as the medullary thick ascending limb and thin limb of the loop of Henle [23]. Previous studies from our laboratory have demonstrated that inner medullary collecting duct production of NO buffers arginine vasopressin-mediated renal medullary vasoconstriction and prevents hypertension in animals in which circulating vasopressin levels are high [15, 17]. Collecting duct-derived NO may also contribute to the acute pressure-natriuresis response. In a recent study, Schneider et al. [25••] demonstrate that NOx excretion is increased in mice in response to acute elevations in renal arterial pressure, but that urinary NOx and sodium excretion are blunted in mice with selective knockout of collecting-duct endothelin 1. These data indicate that endothelin, which is known to stimulate medullary NO production via activation of ETB receptors [26], contributes to the pressure-natriuresis response in mice (Fig. 1). Importantly, mice lacking collecting-duct endothelin display elevated blood pressure, indicating that this pathway contributes to long-term blood pressure control [25••].
Hydrogen Peroxide
In addition to elevated renal outer medullary NOx, Jin et al. [20••] also demonstrated that outer medullary hydrogen peroxide (H2O2) levels increase in parallel with increases in renal arterial pressure. Unlike NOx, however, increases in outer medullary H2O2 levels were blunted following renal decapsulation, suggesting that the increased levels of H2O2 may be secondary to the natriuretic response. The source of H2O2 was not identified in the study by Jin et al. [20••], but O −2 , which can be converted to H2O2 by the action of the enzyme superoxide dismutase (SOD), is known to be produced by the medullary thick ascending limb in response to increased tubular flow and sodium pump activity [27, 28]. As both tubular flow and sodium transport are known to increase when renal perfusion pressure is elevated and both are blunted by renal decapsulation, it is probable that the elevated levels of H2O2 observed were of tubular origin.
Relatively little is known regarding the direct actions of H2O2 on renal medullary tubular and vascular nephron segments. A number of studies in our laboratory have demonstrated that the net effect of H2O2 within the renal medulla is to reduce medullary perfusion and sodium excretion [29, 30]. Therefore, outer medullary H2O2 produced in response to elevated renal perfusion pressure likely acts to dampen the renal natriuretic response. In support of this concept, in a previous study we demonstrated that enhanced production of H2O2 counteracts the vasodilatory and natriuretic actions of infusion of SOD into the renal medulla [31]. These data suggest that both O −2 and H2O2 blunt the natriuretic response to increased renal arterial pressure in vivo. Further studies are required to fully differentiate the actions of O −2 from those of H2O2 at both the whole animal and cellular levels.
Superoxide
In addition to being converted to H2O2 by SOD, O −2 can react directly with a number of molecules to produce hydroxyl radicals, or it can react with NO to form peroxynitrite (OONO−). Relatively little is known regarding the direct actions of hydroxyl radicals or OONO− within the renal medulla; it is thought that much of the effect of O −2 within the renal medulla is mediated by its ability to react with and inactivate NO [3]. In this way, NO and O −2 act as opposing forces altering the redox state of the renal medulla and the set point of the renal pressure-natriuresis curve. There are numerous examples of this yin/yang relationship between NO and O −2 at the cellular level. We have previously demonstrated that O −2 production limits cellular NO availability in perfused medullary thick ascending limb [27]. Further, tubular production of O −2 limits the ability of NO to diffuse through the renal interstitium to the contractile pericyte cell bodies on the descending vasa recta [32]. Therefore, renal medullary O −2 likely modulates renal medullary perfusion and the pressure-natriuresis mechanism through its ability to inactivate NO.
Importantly, a recent study by Hong et al. [33••] demonstrates that most of the reduction of O −2 levels in medullary thick ascending limb in response to increased NO produced by luminal flow occurs via the activation of cellular cGMP and protein kinase G (PKG), not direct scavenging of O −2 by NO to form OONO− (Fig. 1). This finding suggests that, in addition to altered enzymatic pathways of production and scavenging of free radicals, alterations in the renal medullary O −2 and NO balance in disease states may also result from dysfunction of intracellular signaling pathways. Hong et al. [33••] did not investigate the cellular mechanism through which activation of cGMP and PKG reduces tubular O −2 production. However, previous studies suggest that PKG may inhibit NADPH oxidase, the major source of O −2 in medullary thick ascending limb, by preventing translocation of the p47 subunit and activation of the enzyme [34].
O −2 has been demonstrated to increase tubular sodium transport via both Na+/K+/2Cl− (BSC1) and apical Na+/H+ exchangers in medullary thick ascending limb [35, 36]. Both renal medullary oxidative stress [37] and increased tubular transport activity of the medullary thick ascending limb have been associated with a number of models of hypertensive disease, including the Dahl salt-sensitive rat [38], and more recently, high-fat–fed Fischer 344 X Brown Norway rats and rats made hypertensive with angiotensin II [39]. Given the critical role of renal medullary O −2 levels in the control of tubular sodium reabsorption and pressure-natriuresis, much of our current work has focused on identifying the source of excess O −2 in hypertensive disease models. Of particular interest to our own group has been the Dahl salt-sensitive model of hypertension, a genetic rat model that displays many of the traits associated with hypertension in African Americans, such as low renin salt-sensitivity and progressive renal disease [40]. The results from numerous studies in our laboratory indicate that O −2 levels in the renal outer medulla are elevated in Dahl salt-sensitive rats fed 0.4% NaCl when compared with levels in salt-resistant control animals, and that this “oxidative stress” is further exacerbated by high (4.0%) NaCl feeding. Because the thick ascending limb produces more O −2 than any other nephron segment [41], we investigated whether O −2 production was enhanced in outer medullary thick ascending limb from Dahl salt-sensitive rats. When stimulated by incrementing levels of extracellular NaCl in vitro, medullary thick ascending limb from prehypertensive Dahl salt-sensitive rats fed 0.4% NaCl produced more O −2 than medullary thick ascending limb from an identically treated salt-resistant consomic control strain [42•]. Consistent with changes observed in outer medullary oxidative stress in vivo, high NaCl feeding enhanced extracellular NaCl-stimulated O −2 production in medullary thick ascending limb from Dahl salt-sensitive rats [42•]. These data indicated that medullary thick ascending limbs are likely to be the source of excess O −2 in the Dahl salt-sensitive rat model of hypertension.
To investigate the source of excess O −2 production at the cellular level in the Dahl salt-sensitive rat, we recently performed a series of studies on freshly isolated medullary thick ascending limb loaded with either a pH-sensitive dye or a dye sensitive to O −2 . Using fluorescent microscopy techniques to evaluate changes in tubular pH and O −2 production, we investigated whether Na+ or H+ transport was linked to excess O −2 production in Dahl salt-sensitive medullary thick ascending limb [43••]. Our data indicate that a number of stimuli, including cellular shrinkage, angiotensin II, and acid loading, stimulated O −2 production in medullary thick ascending limb through a common amiloride-sensitive pathway that is linked to cellular H+ efflux and is augmented in the Dahl salt-sensitive rat model [43••]. Numerous lines of evidence indicate that this H+ efflux pathway is independent of sodium transport and is mediated by an as yet unidentified H+ transporter, not the classic target of amiloride in medullary thick ascending limb, the Na+/H+ exchanger [43••]. Identification of this unknown amiloride-sensitive H+ transporter may be key in identifying the molecular source of renal medullary oxidative stress and dysfunctional pressure-natriuresis in Dahl salt-sensitive rats.
Hypoxia, Renal Medullary Reactive Oxygen Species, and Pressure-Natriuresis
The renal outer medulla is thought to operate on the verge of hypoxia even under normal physiological conditions because of the heavily respiring medullary thick ascending limb (responsible for reabsorbing about 25% of the filtered sodium load) and a relatively low rate of local perfusion when compared with the renal cortex [44]. Alterations in the balance of medullary reactive oxygen and nitrogen species can promote local hypoxia [44]. NO, in addition to reducing tubular sodium transport [45] and increasing medullary blood flow [12], has direct effects on the mitochondria to limit respiration and O2 use, thereby acting to increase local pO2 [46]. Conversely, O −2 opposes the actions of NO and acts to reduce local oxygen levels in vivo [3]. It may not be surprising, then, that reduced medullary tissue pO2 is a common finding in hypertensive animal models that exhibit renal oxidative stress [47]. Reduced renal tissue pO2 has been observed in spontaneously hypertensive rats (SHR), angiotensin II–infused rats, and the two-kidney/one-clip rat model of hypertension [47]. Importantly, the reduced pO2 observed in these models appears to be related to enhanced oxidative stress and reduced bioavailability of NO. Treatment with the superoxide scavenger Tempol restores renal tissue oxygenation in SHR to levels observed in normotensive Wistar-Kyoto (WKY) rats, whereas L-NAME reduces tissue pO2 in WKY rats but not in SHR [48]. These data indicate that O −2 acts to reduce tissue pO2 in SHR at least in part by inactivating NO.
Renal medullary hypoxia may play a critical role in preventing the development of renal oxidative stress and hypertension. The production of O −2 by NADPH oxidase is limited at physiologic pO2 levels less than 30 mm Hg (the normal operating range of pO2 within the renal medulla) [49]. Because NADPH oxidase is a significant source of O −2 in the renal outer medulla [37] and increased levels of O −2 reduce local pO2, medullary oxidative stress may be somewhat self-limiting by reducing the bioavailability of oxygen as a substrate for NADPH oxidase. Perhaps even more interesting is a recent observation by Li et al. [50••] demonstrating the importance of hypoxia inducible factor (HIF)-1α signaling within the renal medulla in salt-sensitive hypertension. Renal medullary specific inhibition of HIF-1α in normally salt-resistant Sprague-Dawley rats with targeted oligonucleotides resulted in the development of salt-sensitive hypertension in these animals. Further, the natriuretic response to acute intravenous loading of NaCl was blunted in HIF-1α–inactivated animals [50••]. These findings demonstrate the importance of a normal hypoxic signaling pathway within the renal medullary region for the maintenance of normal levels of arterial pressure. As HIF-1α, a transcription factor that is activated by cellular hypoxia, regulates many oxygen-sensitive genes including activation of NO synthase, it is likely that HIF-mediated hypoxic signaling acts to modulate medullary reactive oxygen and nitrogen species and the pressure-natriuresis mechanism. Therefore, hypoxia may be a key mediator of renal medullary reactive oxygen and nitrogen species, whereby dysfunction in hypoxic signaling may play a role in the development of renal oxidative stress and hypertension.
Conclusions
The renal pressure-natriuresis mechanism is the dominant controller of body fluid balance and long-term arterial pressure. In recent years, it has become clear that the balance of reactive oxygen and nitrogen species within the renal medullary region is a key determinant of the set point of the renal pressure-natriuresis curve. The development of renal medullary oxidative stress causes dysfunction of the pressure-natriuresis mechanism and contributes to the development of hypertension in numerous disease models. Among notable recent advances in this field are the identification of collecting-duct endothelin signaling as a key contributor to the NO-mediated pressure-natriuresis response in mice [25••], the characterization of amiloride-sensitive H+ transport as a major source of oxidative stress in the renal outer medulla of Dahl salt-sensitive rats [43], identification of NO-induced cGMP/PKG as a signaling pathway to reduce O −2 formation in medullary thick ascending limb [33••], and the demonstration of the importance of HIF-1α signaling in preventing salt-sensitive hypertension [50]. Identifying the underlying causes of renal oxidative stress in animal models of hypertension will undoubtedly add to our understanding of the pathogenesis of this disease and may provide novel approaches to its treatment.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Guyton AC: Dominant role of the kidneys and accessory role of whole-body autoregulation in the pathogenesis of hypertension. Am J Hypertens 1989, 2(7):575–585.
Cowley AW Jr, Mattson DL, Lu S, Roman RJ: The renal medulla and hypertension. Hypertension 1995, 25(4 Pt 2):663–673.
Cowley AW Jr: Renal medullary oxidative stress, pressure-natriuresis, and hypertension. Hypertension 2008, 52(5):777–786.
Evans RG, Majid DS, Eppel GA: Mechanisms mediating pressure natriuresis: what we know and what we need to find out. Clin Exp Pharmacol Physiol 2005, 32(5–6):400–409.
Cowley AW, Roman RJ, Fenoy FJ, Mattson DL: Effect of renal medullary circulation on arterial pressure. J Hypertens Suppl 1992, 10(7):S187–S193.
Garcia-Estan J, Roman RJ: Role of renal interstitial hydrostatic pressure in the pressure diuresis response. Am J Physiol 1989, 256(1 Pt 2):F63–F70.
Williams JM, Sarkis A, Lopez B, et al.: Elevations in renal interstitial hydrostatic pressure and 20-hydroxyeicosatetraenoic acid contribute to pressure natriuresis. Hypertension 2007, 49(3):687–694.
Zhang YB, Magyar CE, Holstein-Rathlou NH, McDonough AA: The cytochrome P-450 inhibitor cobalt chloride prevents inhibition of renal Na,K-ATPase and redistribution of apical NHE-3 during acute hypertension. J Am Soc Nephrol 1998, 9(4):531–537.
Cowley AW Jr: Long-term control of arterial blood pressure. Physiol Rev 1992, 72(1):231–300.
Majid DS, Navar LG: Nitric oxide in the mediation of pressure natriuresis. Clin Exp Pharmacol Physiol 1997, 24(8):595–599.
Guarasci GR, Kline RL: Pressure natriuresis following acute and chronic inhibition of nitric oxide synthase in rats. Am J Physiol 1996, 270(2 Pt 2):R469–R478.
Mattson DL, Roman RJ, Cowley AW Jr: Role of nitric oxide in renal papillary blood flow and sodium excretion. Hypertension 1992, 19(6 Pt 2):766–769.
Mattson DL, Lu S, Nakanishi K, et al.: Effect of chronic renal medullary nitric oxide inhibition on blood pressure. Am J Physiol 1994, 266(5 Pt 2):H1918–H1926.
Szentivanyi M Jr, Zou AP, Mattson DL, et al.: Renal medullary nitric oxide deficit of Dahl S rats enhances hypertensive actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol 2002, 283(1):R266–R272.
O’Connor PM, Cowley AW Jr: Vasopressin-induced nitric oxide production in rat inner medullary collecting duct is dependent on V2 receptor activation of the phosphoinositide pathway. Am J Physiol Renal Physiol 2007, 293(2):F526–532.
Szentivanyi M Jr, Maeda CY, Cowley AW Jr: Local renal medullary L-NAME infusion enhances the effect of long-term angiotensin II treatment. Hypertension 1999, 33(1 Pt 2):440–445.
Park F, Zou AP, Cowley AW Jr: Arginine vasopressin-mediated stimulation of nitric oxide within the rat renal medulla. Hypertension 1998, 32(5):896–901.
Yuan B, Cowley AW Jr: Evidence that reduced renal medullary nitric oxide synthase activity of dahl s rats enables small elevations of arginine vasopressin to produce sustained hypertension. Hypertension 2001, 37(2 Part 2):524–528.
Fenoy FJ, Ferrer P, Carbonell L, Garcia-Salom M: Role of nitric oxide on papillary blood flow and pressure natriuresis. Hypertension 1995, 25(3):408–414.
•• Jin C, Hu C, Polichnowski A, et al.: Effects of renal perfusion pressure on renal medullary hydrogen peroxide and nitric oxide production. Hypertension 2009, 53(6):1048–1053. Using in vivo microdialysis techniques, Jin et al. demonstrate that increasing renal perfusion pressure stimulates renal medullary NO and H 2 O 2 . Importantly, NO stimulation was not affected by renal decapsulation and inhibition of the renal natriuretic response, suggesting that the increased production of NO may be a primary event in the renal pressure-natriuresis cascade.
Ortiz PA, Hong NJ, Garvin JL: NO decreases thick ascending limb chloride absorption by reducing Na(+)-K(+)-2Cl(-) cotransporter activity. Am J Physiol Renal Physiol 2001, 281(5):F819–F825.
• Lieb DC, Kemp BA, Howell NL, et al.: Reinforcing feedback loop of renal cyclic guanosine 3′5′-monophosphate and interstitial hydrostatic pressure in pressure-natriuresis. Hypertension 2009, 54(6):1278–1283. Lieb et al. demonstrate that cyclic guanosine 3′5′-monophosphate is produced in response to increases in RIHP and also acts to increase RIHP through an unknown mechanism. By activating a positive feedback loop, cyclic guanosine 3′5′-monophosphate may allow the kidney to rapidly reduce sodium and water reabsorption when renal perfusion pressure is elevated.
Wu F, Park F, Cowley AW Jr, Mattson DL: Quantification of nitric oxide synthase activity in microdissected segments of the rat kidney. Am J Physiol 1999, 276(6 Pt 2):F874–F881.
Zhang Z, Pallone TL: Response of descending vasa recta to luminal pressure. Am J Physiol Renal Physiol 2004, 287(3):F535–F542.
•• Schneider MP, Ge Y, Pollock DM, et al.: Collecting duct-derived endothelin regulates arterial pressure and Na excretion via nitric oxide. Hypertension 2008, 51(6):1605–1610. Schneider et al. demonstrate that that mice lacking collecting-duct endothelin display elevated blood pressure and an altered pressure-natriuresis response, indicating that this pathway contributes to long-term blood pressure control.
Nakano D, Pollock JS, Pollock DM: Renal medullary ETB receptors produce diuresis and natriuresis via NOS1. Am J Physiol Renal Physiol 2008, 294(5):F1205–F1211.
Abe M, O’Connor P, Kaldunski M, et al.: Effect of sodium delivery on superoxide and nitric oxide in the medullary thick ascending limb. Am J Physiol Renal Physiol 2006, 291(2):F350–F357.
Hong NJ, Garvin JL: Flow increases superoxide production by NADPH oxidase via activation of Na-K-2Cl cotransport and mechanical stress in thick ascending limbs. Am J Physiol Renal Physiol 2007, 292(3):F993–F998.
Makino A, Skelton MM, Zou AP, et al.: Increased renal medullary oxidative stress produces hypertension. Hypertension 2002, 39(2 Pt 2):667–672.
Taylor NE, Cowley AW Jr: Effect of renal medullary H2O2 on salt-induced hypertension and renal injury. Am J Physiol Regul Integr Comp Physiol 2005, 289(6):R1573–R1579.
Chen YF, Cowley AW Jr, Zou AP: Increased H(2)O(2) counteracts the vasodilator and natriuretic effects of superoxide dismutation by tempol in renal medulla. Am J Physiol Regul Integr Comp Physiol 2003, 285(4):R827–R833.
Mori T, O’Connor PM, Abe M, Cowley AW Jr: Enhanced superoxide production in renal outer medulla of Dahl salt-sensitive rats reduces nitric oxide tubular-vascular cross-talk. Hypertension 2007, 49(6):1336–1341.
•• Hong NJ, Garvin JL: Nitric oxide reduces flow-induced superoxide production via cGMP-dependent protein kinase in thick ascending limbs. Am J Physiol Renal Physiol 2009, 296(5):F1061–F1066. Hong et al. demonstrate that most of the reduction of O 2 − levels in medullary thick ascending limb in response to increased NO produced by luminal flow occurs via the activation of cellular cGMP and protein kinase G (PKG), not direct scavenging of O 2 − by NO to form OONO −.
Muzaffar S, Shukla N, Bond M, et al.: Acute inhibition of superoxide formation and Rac1 activation by nitric oxide and iloprost in human vascular smooth muscle cells in response to the thromboxane A2 analogue, U46619. Prostaglandins Leukot Essent Fatty Acids 2008, 78(4–5):247–255.
Juncos R, Garvin JL: Superoxide enhances Na-K-2Cl cotransporter activity in the thick ascending limb. Am J Physiol Renal Physiol 2005, 288(5):F982–F987.
Juncos R, Hong NJ, Garvin JL: Differential effects of superoxide on luminal and basolateral Na+/H + exchange in the thick ascending limb. Am J Physiol Regul Integr Comp Physiol 2006, 290(1):R79–R83.
Taylor NE, Glocka P, Liang M, Cowley AW Jr: NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S rats. Hypertension 2006, 47(4):692–698.
Garcia NH, Plato CF, Stoos BA, Garvin JL: Nitric oxide-induced inhibition of transport by thick ascending limbs from Dahl salt-sensitive rats. Hypertension 1999, 34(3):508–513.
Riazi S, Tiwari S, Sharma N, et al.: Abundance of the Na-K-2Cl cotransporter NKCC2 is increased by high-fat feeding in Fischer 344 X Brown Norway (F1) rats. Am J Physiol Renal Physiol 2009, 296(4):F762–F770.
Kurtz TW: Genetic models of hypertension. Lancet 1994, 344(8916):167–168.
Li N, Yi FX, Spurrier JL, et al.: Production of superoxide through NADH oxidase in thick ascending limb of Henle’s loop in rat kidney. Am J Physiol Renal Physiol 2002, 282(6):F1111–F1119.
• O’Connor PM, Lu L, Schreck C, Cowley AW Jr: Enhanced amiloride-sensitive superoxide production in renal medullary thick ascending limb of Dahl salt-sensitive rats. Am J Physiol Renal Physiol 2008, 295(3):F726–F733. The authors demonstrate that O 2 − production is enhanced in medullary thick ascending limb from Dahl salt-sensitive rats and that this enhanced O 2 − production is sensitive to inhibition by amiloride analogues.
•• O’Connor PM, Lu L, Liang M, Cowley AW Jr: A novel amiloride-sensitive H+ transport pathway mediates enhanced superoxide production in thick ascending limb of salt-sensitive rats, not Na+/H+ exchange. Hypertension 2009, 54(2):248–254. In this follow-up study, the authors demonstrate that excess amiloride-sensitive O 2 − production observed in medullary thick ascending limb of Dahl salt-sensitive rats is related to activation of a novel H + transport pathway, a pathway that is upregulated in the Dahl salt-sensitive rat.
O’Connor PM: Renal oxygen delivery: matching delivery to metabolic demand. Clin Exp Pharmacol Physiol 2006, 33(10):961–967.
Herrera M, Ortiz PA, Garvin JL: Regulation of thick ascending limb transport: role of nitric oxide. Am J Physiol Renal Physiol 2006, 290(6):F1279–F1284.
Brown GC: Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett 1995, 369(2–3):136–139.
Welch WJ: Intrarenal oxygen and hypertension. Clin Exp Pharmacol Physiol 2006, 33(10):1002–1005.
Welch WJ, Blau J, Xie H, et al.: Angiotensin-induced defects in renal oxygenation: role of oxidative stress. Am J Physiol Heart Circ Physiol 2005, 288(1):H22–H28.
Chen Y, Gill PS, Welch WJ: Oxygen availability limits renal NADPH-dependent superoxide production. Am J Physiol Renal Physiol 2005, 289(4):F749–F753.
•• Li N, Chen L, Yi F, et al.: Salt-sensitive hypertension induced by decoy of transcription factor hypoxia-inducible factor-1alpha in the renal medulla. Circ Res 2008, 102(9):1101–1108. Li et al. demonstrate the importance of HIF signaling in the maintenance of blood pressure in rats fed high-sodium diets. Inhibition of HIF signaling specifically in the renal medulla resulted in marked salt sensitivity of blood pressure in otherwise normotensive, salt-resistant Sprague-Dawley rats.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
O’Connor, P.M., Cowley, A.W. Modulation of Pressure-Natriuresis by Renal Medullary Reactive Oxygen Species and Nitric Oxide. Curr Hypertens Rep 12, 86–92 (2010). https://doi.org/10.1007/s11906-010-0094-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-010-0094-6