
Abstract
While mortality rates related to cardiovascular disease (CVD) have decreased over time among adults with HIV, excess risk of CVD in the HIV-infected population may persist despite highly active antiretroviral therapy (HAART) treatment and aggressive CVD risk factor control. Beyond atherosclerotic CVD, recent studies suggest that HIV infection may be associated with left ventricular systolic and diastolic function, interstitial myocardial fibrosis, and increased cardiac fat infiltration. Thus, with the increasing average age of the HIV-infected population, heart failure and arrhythmic disorders may soon rival coronary artery disease as the most prevalent forms of CVD. Finally, the question of whether HIV infection should be considered in clinical risk stratification has never been resolved, and this question has assumed new importance with recent changes to lipid treatment guidelines for prevention of CVD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
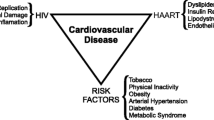
The success of antiretroviral therapy (ART) has been transformational in the lives of people living with HIV infection since it became available in the mid-1990s. Survival is markedly better than in the pre-ART era [1, 2], and some studies now suggest that people on ART have life expectancy approaching that of HIV-uninfected individuals [3]. At the same time, ever since effective ART was introduced, there have been questions about the potential effects of long-term-treated HIV infection on the development of cardiovascular disease (CVD). These concerns continue to persist, even as HIV treatment is refined with fewer metabolic side effects, better adherence, more convenience, and earlier initiation of uninterrupted therapy [4, 5]. The issue is further complicated by the high prevalence of traditional CVD risk factors among many HIV-infected individuals, including cigarette smoking [6], and the effects that chronic immune activation and inflammation may have on developing CVD risk [7].
We begin this up-to-date review by describing data during the highly active antiretroviral therapy (HAART) era on mortality trends in the HIV-infected population. Next, we review studies that have addressed whether elevated CVD risk persists in individuals on modern, successful ART. Most often in the past, the focus of CVD research in the HIV setting has been on atherosclerotic vascular disease processes, as has been reviewed elsewhere [8, 9]. In the subsequent section of this review, we describe the mechanisms of cardiac structural and functional abnormalities that may be important among ART-treated patients, possibly foreshadowing a future epidemic of heart failure and arrhythmic disease in HIV-infected adults. Finally, we describe recent changes to the clinical guidelines concerning the use of cholesterol-lowering drugs. Lipid-lowering treatments are perhaps the most widely relevant tool for CVD prevention, and the new lipid guidelines have implications for clinical care and research in the HIV-infected population.
Trends in Mortality Show Continued Importance of CVD in the HIV-Infected Population
Mortality trends show population-wide shifts in the conditions that are most likely to lead to death in the HIV-infected population and how these conditions may differ from those affecting the general population. It is well-known that among the US population on the whole, the age-adjusted mortality rate due to coronary heart disease has declined steadily over the past 50 years [10]. These improvements have been attributed to both greater awareness and treatment of CVD risk factors leading to primary prevention, and the availability of pharmacological and surgical interventions leading to secondary prevention [11]. Understanding recent trends in CVD mortality in the HIV-infected population is of great interest [12, 13••, 14] given the unique circumstances of HIV infection and the modern context of effective and widely available ART.
The multicohort D:A:D study found that among 49,731 study participants seen at clinics in Europe, the USA, and Australia, the age-standardized CVD mortality rate decreased from 2.0/1000 person-years in 1999 to 0.7/1000 person-years in 2011 [13••]. Decreasing CVD mortality rates were also found among those with HIV viral loads below 50 copies per ml, falling from 1.4/1000 to 0.7/1000. Our group analyzed surveillance data and vital statistics records from 145,845 New York City residents living with HIV between 2001 and 2012 [14]. This suggested a decrease in the age-standardized CVD mortality rate, from 5.1/1000 in 2001 to 2.7/1000 in 2012. Compared with the D:A:D collaboration, the New York City study had higher absolute CVD mortality rates, possibly due to the use of city-wide surveillance data to capture a diverse area-based sample. Nonetheless, despite this difference, both the New York City study and the D:A:D collaboration documented approximately a 50 % decrease in the CVD mortality rate over the last decade among adults with HIV infection.
Despite decreases in CVD mortality rates, non-AIDS-defining conditions constitute an ever-growing proportion of deaths in the HIV-infected population owing to the continued fall in deaths due to AIDS. Numerous studies in different populations have found that CVD largely contributed to the increase in the relative importance of non-AIDS death to the overall mortality rate [15–18].
Two recent studies, one from the SMART and ESPRIT clinical trials and the other from the European COHERE collaboration, have suggested that people with HIV infection who are well-controlled on ART have mortality rates close to that observed among the general population [19•, 20]. In contrast, our study comparing HIV-infected New Yorkers to the general population found that those with suppressed viral loads (defined as plasma HIV RNA <400 copies per ml) still had a 53 % higher rate of CVD mortality as compared with uninfected New Yorkers after controlling for sex, age, race/ethnicity, and county of residence [14]. Thus, in some demographic groups with HIV infection, increased CVD risk may persist despite virologic suppression. How much of this elevated risk can be attributed to underlying CVD risk factors, to accelerated aging of the immune or cardiovascular systems, or to disease processes that occur despite suppression of plasma HIV RNA levels is the subject of active investigation.
Lessons Learned From Contemporary Studies of Incident CVD in the HIV-Infected Population
Studies continue to address the incidence and risk factors for incident CVD events in the HIV-infected population. The most valuable such studies are those that document the degree of excess CVD risk among those with HIV as compared with HIV-uninfected comparison groups. Contemporary studies are also important because of dramatic changes over the past decade in HIV treatment strategies. One such example was reported from the Veterans Aging Cohort Study Virtual Cohort, which accumulated 363 acute myocardial infarction (MI) events among over 27,000 HIV-infected veterans between 2003 and 2009 [21••]. A comparison group of 55,000 HIV-uninfected veterans with 508 acute MI events was also studied. This group was matched to HIV-infected individuals by age, race/ethnicity, clinical site, and calendar year. To arrive at this case group, the investigators first excluded 17 % of participants in their database who had ICD-9 codes for cardiovascular diagnoses, events, and procedures prior to study baseline. Among MI events that occurred over 6 years of follow up, 61.3 % occurred at Veterans’ Administration facilities and were subject to adjudication by medical records review. An additional 18.5 % of events that occurred outside the Veterans’ Administration system were identified by Medicare inpatient ICD-9 code 410.XX, while 20.2 % of events were coronary deaths. After adjusting for traditional CVD risk factors, comorbidities, and substance use, HIV-infected veterans had an increased risk of incident MI compared with uninfected veterans (hazard ratio [HR], 1.48; 95 % confidence interval [CI], 1.27–1.72).
Similar findings were reported from the Kaiser Permanente Northern California clinical population. Updating an earlier study from 2002 [22••], Klein and colleagues reported an average of 5.8 years of follow-up among 24,768 HIV-infected and 257,600 HIV-uninfected patients enrolled in their system during 1996 through 2011 [23]. HIV-infected and HIV-uninfected groups were frequency-matched to each other by year, age, sex, and medical center. Discharge diagnosis codes (ICD-9 codes 410.XX) were used to identify 320 MIs among HIV-infected individuals and 2483 MIs among HIV-uninfected controls. The adjusted relative risk (RR) of incident MI was 1.4 (95 % CI 1.2–1.6) comparing the HIV-infected group versus the HIV-uninfected group. The pattern of results over time suggested that relative risk in the HIV-infected population declined from a nearly two-fold excess in the early HAART era (HR = 1.8, 95 % CI 1.3–2.6 during 1996–1999) to a small or null risk in the most recent years (HR = 1.0, 95 % CI 0.7–1.4 during 2010–2011). A high prevalence of statin use in the latter years of the study might explain the diminishment over time of the HIV-related relative risk.
Several aspects of design and clinical characteristics distinguish the two abovementioned studies. Only the Veterans’ Administration study obtained adjudication of medical records to confirm MI events. As compared with the Kaiser Permanente cohort, in the veterans’ study, the prevalence of substance abuse was higher, and this important confounder was better matched between HIV-infected and HIV-uninfected groups. The population of HIV-infected veterans was nearly half African American, while the largest race-ethnic groups in the Kaiser Permanente population were whites and Hispanics. Despite these differences, the Veterans’ Administration and Kaiser Permanente studies estimated a very similar overall HR for incident MI of 1.4 to 1.5 associated with HIV-positive status. These were both well-conducted studies that obtained large sample sizes, that were restricted to comprehensive health care provider organizations, thus reducing ascertainment and information biases, and that extended into the contemporary HAART era. On the other hand, as in most observational studies of CVD among the HIV-infected population, it is difficult to exclude potential threats to validity. Providers may have heightened awareness of CVD risk in patients with HIV infection, which potentially may lead to different thresholds for starting CVD-reducing treatments or referring patients for suspected CVD if HIV infection is present. There may also be survival effects in the older HIV-infected population, such that those with the greatest socioeconomic adversity, major non-HIV comorbidities, and propensity for treatment non-adherence may have been depleted from the population over time due to high mortality. These biases may lead to either overestimation or underestimation of the risk of CVD among persons with HIV infection. Finally, both the Kaiser Permanente and the Veterans’ Administration study included few females.
Another important methodological issue was brought to light by the CNICS group’s 2014 report on ascertainment and classification of CVD events in the HIV-infected population [24•]. Based upon careful medical records review, this well-conducted investigation concluded that approximately half of MI events recorded among HIV patients from eight US clinical sites were secondary MIs. These secondary forms of MI were caused by atypical mechanisms such as vasospasm from use of cocaine or sepsis, which can precipitate an acute CVD event above and beyond traditional CVD risk factors. As compared with those sustaining primary MI events, patients with secondary MI were, on average, sicker and had lower CD4+ T cell counts. Sabin and colleagues concluded that even with protocol-driven endpoint validation in the D:A:D collaboration, a preponderance of apparent cerebrovascular events in individuals with low CD4+ T cell counts may have been related to HIV-associated central nervous system disorders rather than traditional mechanisms of stroke [25]. Thus, in order to identify the most effective CVD prevention strategies, the relative importance of traditional atherosclerotic pathophysiological processes versus other contributing mechanisms of CVD in the HIV-patient population needs to be clarified.
What Is the Cardiovascular Risk Status Under Conditions of Successful ART?
Current clinical guidelines now aim for early and aggressive ART. Therefore, the risk of CVD in patients maintained on suppressive ART has become an increasingly important question. Among the most important present-day clinical treatment goals are consistent suppression of plasma viremia below detectable limits and initiation of ART at high circulating CD4+ T cell counts. The risk of incident MI among individuals with these favorable HIV clinical characteristics was examined in several recent studies including those reviewed above as well as others.
In the Kaiser Permanente Northern California study, as compared with HIV-uninfected controls, HIV-infected patients with greater degree of CD4+ T cell depletion had progressively higher risk of MI. In contrast, among HIV-infected subjects with CD4 ≥500 cells per microliter (ml), no statistically significant increase in risk of MI was observed comparing versus HIV-uninfected subjects (RR = 1.18, 95 % CI 0.96–1.45). In the Veterans’ Administration study, lower CD4+ T cell count was also associated with more unfavorable risk of incident MI in the HIV-infected group. The HR was 1.43 (95 % CI 1.21–1.69) comparing HIV-infected patients with CD4+ T cell count of 200 cells per ml or greater versus HIV-uninfected controls. Unfortunately, in the Veterans’ Administration study, comparisons between those having higher CD4 T cell counts ( ≥ 500 cells per ml) versus HIV-uninfected were not presented.
Circulating HIV RNA levels also were examined in relation to MI risk in both the Kaiser Permanente and Veterans’ Administration studies. The Kaiser Permanente study found an increased HR for incident MI among those both above and below the threshold of HIV RNA of 500 copies per ml (comparing HIV-infected patients with HIV RNA <500 copies per ml versus HIV-uninfected, HR = 1.41, 95 % CI 1.21–1.64). Similarly, in comparison with HIV-uninfected veterans, HIV-infected veterans with HIV RNA below 500 copies per ml had an HR of 1.39 (95 % CI 1.17–1.66). This was lower than the HR observed among veterans with HIV RNA above 500 copies per ml (HR = 1.75), yet both high and low HIV RNA groups had elevated risk of MI versus controls.
These recent studies suggest that clinical HIV disease status measures including CD4+ T cell count and viral load can identify HIV-infected individuals at highest risk of CVD, confirming prior studies that lacked HIV-uninfected comparators [25–27]. A common limitation has been the use of HIV RNA detection thresholds that are higher than the more sensitive tests for measuring plasma viremia currently in clinical use (e.g., HIV RNA <40 copies per ml). To address these limitations, our Women’s Interagency HIV Study (WIHS) and Multicenter AIDS Cohort Study (MACS) investigations used carotid artery B mode ultrasound to examine progression of subclinical atherosclerosis in a group of HIV-infected individuals with persistent virologic suppression [28••]. Persistent viral suppression was defined by protocol-driven measurements of plasma HIV RNA (viral load) levels <80 copies per ml (WIHS) or <50 copies per ml (MACS) during up to 16 semiannual measurements. The criterion for persistent viral suppression allowed for 1 virologic “blip” during the follow-up period as long as it was below 500 copies per ml, but otherwise all HIV-infected participants were documented to have excellent viral suppression during the 7–8 year period of repeated carotid artery ultrasound measurements. Among 199 HIV-infected participants who were receiving ART and persistently virologically suppressed, we observed an increased risk of new focal carotid artery plaque formation compared with the HIV-uninfected group (adjusted RR = 1.77, 95 % CI 1.13–2.77). As a study of subclinical atherosclerosis rather than incident CVD, this report has limitations associated with the use of a surrogate endpoint. On the other hand, the WIHS/MACS collaboration is one of the few prospective studies that have characterized HIV disease parameters using frequent measurements over time, rather than only baseline, recent, peak, or nadir values of CD4+ T cell counts or HIV RNA.
In summary, several recent studies point to the possibility that even with current antiretroviral treatment approaches, suppressing plasma HIV RNA may not control increased risk of CVD in adults with longstanding HIV infection. Therefore, this suggests the need to study mechanisms of increased CVD that persist even in ideally treated HIV-infected patients. Candidate mechanisms that may trigger vascular dysfunction or hypercoagulability may include inflammation driven by persistent translocation of bacterial species in the gut, irreversible immune damage sustained in primary HIV infection, and liver fibrosis due to hepatitis virus infection or other hepatic disease. In addition, as discussed in the next section, the hypothesis that there may be direct cardiac effects of HIV infection has recently been supported by myocardial imaging and biomarker studies conducted during in the HAART era.
Beyond Atherosclerosis: the Potential Role of Cardiac Damage in HIV for Risk of Heart Failure and Dysrhythmias
While much of the work to date has focused upon coronary artery disease and other measures related to atherosclerosis burden, the impact of HIV infection and its treatment on myocardial structure and function has been an area of resurgent interest. Prior to the advent of combination ART, dilated cardiomyopathy was a striking complication of late-stage HIV infection. Associated with severe systolic heart failure (HF) and poor prognosis, dilated cardiomyopathy had a prevalence as high as 30 % in some studies [29, 30]. The disorder appeared to be related to possible myocardial or dendritic cell invasion by HIV itself or other viral pathogens, as well as inflammation and autoimmunity [29].
Dilated cardiomyopathy is no longer a prominent manifestation of HIV infection in the HAART era. Accumulating data suggest, however, that such severe HIV-associated cardiomyopathy has been replaced by a more insidious form, one where mild left ventricular (LV) systolic dysfunction is common and LV diastolic dysfunction is pronounced [31]. A recent meta-analysis arrived at pooled prevalences of LV systolic and diastolic dysfunction in HIV-infected persons of 8.3 and 43.4 %, respectively [31]. These frequencies of subclinical LV dysfunction are striking given that the mean age of participants studied was 41 years old. As compared with population-based estimates, the prevalence of reduced LV ejection fraction is about threefold higher, and abnormal LV diastolic function two- to threefold greater, in HIV-infected individuals than that reported in older white [32] or African American [33, 34] non-HIV cohorts. Such figures are a cause for concern, because they suggest that as the HIV-infected population continues to age—with projections that over half of HIV-infected individuals will be 50 or older by 2020 [35]—there may be an upsurge in HF on the horizon. This is all the more worrisome because frequent comorbidities of HIV infection, including lung disease, chronic kidney disease, and anemia, may contribute to and exacerbate HF symptoms in the absence of severe LV systolic dysfunction [36, 37]. Recent data from US veterans showed HIV infection to be associated with an almost twofold risk of incident HF in the absence of previously documented coronary heart disease (CHD) [38•]. No less important, myocardial dysfunction sets up a substrate for ventricular dysrhythmias, and offers a link to the elevated risk of atrial fibrillation/flutter [39] sudden cardiac death reported in association with HIV infection [40].
The determinants of HIV-associated myocardial dysfunction in the ART era are likely multifactorial [41], but remain incompletely delineated. Persistent viral replication has been implicated, and HIV viremia appeared to be a leading predictor of incident HF in the male veteran population [38•]. Regarding subclinical ventricular dysfunction, residual HIV-associated inflammation has been linked to both systolic and diastolic abnormalities. Older age and hypertension are additional risk factors for diastolic dysfunction, while smoking and prior myocardial infarction are determinants of systolic dysfunction [31].
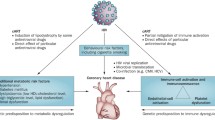
With the introduction of combination ART and successful long-term suppression of HIV, direct myocardial assault by HIV-1 and cardiotropic viruses has been markedly diminished, but likely not eliminated. In particular, persistence of HIV-1 within reservoirs during combination ART use, along with viral co-infections and translocation of intestinal bacteria, may foster a state of chronic immune activation and inflammation, which heightens oxidative stress and results in endothelial dysfunction [42]. This can lead to cardiomyocyte dysfunction and increased stiffness, and eventuate in heightened interstitial fibrosis [43]. Apart from pro-inflammatory insults from HIV and co-infections, illicit drugs, such as cocaine [44], opiates [45], and alcohol [46], can play a major role.
Furthermore, ART-related effects are also potential contributors to myocardial dysfunction [47]. Nucleoside reverse transcriptase inhibitors (NRTI) such as zidovudine and stavudine promote lipodystrophy and alter glucose metabolism, and have been linked to LV dysfunction through associated mitochondrial toxicity in some, albeit not all, studies [30]. As for newer NRTIs with more favorable side effect profiles, abacavir has been associated with increased CVD risk in several studies [48, 49], but this has not been consistently confirmed [50]. Tenofovir has been linked to greater risk of HF, though this finding requires replication [49]. Protease inhibitors (PIs), particularly indinavir and ritonavir-boosted lopinavir, promote lipodystrophy by inhibiting adipocyte differentiation, and foster both dyslipidemia and insulin resistance. While ART lowers overall CHD risk [51], it still remains possible that specific PIs present higher CVD risk than other agents [52, 53, 54]. By contrast, the newer PI atazanavir, which has less adverse metabolic effects, has not been linked to increased CHD risk [55]. Overall, however, the extent to which the metabolic effects of ART contribute to HIV-associated cardiac dysfunction is not well defined.
A hallmark of cardiac aging is increased myocardial fibrosis [56], which heightens cardiac stiffness and can eventuate in HF with preserved ejection fraction (HFpEF) [57]. Such diffuse interstitial fibrosis is distinct from the replacement fibrosis (scar) that occurs with different degrees of transmurality following myocardial infarction from coronary atherothrombosis [58]. Like replacement fibrosis, diffuse interstitial fibrosis can be non-invasively quantitated by cardiac magnetic resonance (CMR) imaging techniques [58]. CMR T1 mapping [59], in particular, has been applied in a general population setting to show that diffuse interstitial fibrosis increases with age [60] and is associated with hypertension [61]. Apart from fibrosis, experimental studies have shown that mismatch between cardiomyocyte uptake and oxidation of free fatty acids leads to cardiomyocyte accumulation of triglycerides and other lipids [62]. This situation supervenes in disorders of glucose metabolism [63] or increased cardiac stress [64]. Clinical use of [1] H magnetic resonance spectroscopy (MRS) has allowed identification of high intramyocardial triglyceride content in obesity and insulin resistance/diabetes [65, 66], as well as older age [67] and severe aortic stenosis [68]. Such studies have documented a correlation between myocardial triglyceride level and LV dysfunction [69–71], although whether this relationship is causal (lipotoxicity) or reflects common upstream processes remains uncertain [63].
Evidence that cardiac fibrosis and steatosis may underlie HIV-related cardiac dysfunction has recently emerged. A recent study using CMR imaging found increased LV patchy fibrosis by late gadolinium enhancement and fat content by MRS, along with depressed measures of myocardial deformation, in HIV-infected patients receiving combination ART as compared with age-matched healthy controls [72••]. The patchy fibrosis occurred in 76 % of HIV-infected patients (as compared with 13 % of controls, p < 0.001), primarily in the middle and subepicardial layers of the basal inferolateral wall, a pattern consistent with prior myocarditis. A second study has reported similar findings, detecting impaired LV strain, greater diffuse myocardial fibrosis (this time by CMR T1 mapping), and higher myocardial lipid content in ART-treated HIV-infected patients versus age-, sex-, and race-matched healthy HIV-uninfected controls [73]. Consistent results were reported for LV strain and myocardial fat content in a third, smaller case–control study [74]. Myocardial infarcts were detected infrequently in these studies, but this owed to their design in excluding clinically overt CHD. Together, these findings support the role of diffuse myocardial fibrosis and lipid deposition in HIV-associated subclinical LV dysfunction. These modest-sized cross-sectional studies, however, relied on healthy HIV-uninfected volunteers as controls, which prevent distinguishing the influence of HIV infection itself, its treatment, and associated risk factors on the differences in LV function or myocardial tissue composition observed. Larger studies with better-matched HIV-uninfected controls and more detailed characterization of exposures are necessary to determine the true extent and determinants of such differences.
Defining the Role of Statins in the HIV-Infected Patient Population
As noted above, among the most important gaps in the literature is the lack of high-quality data describing absolute and relative risks of CVD among HIV patients, especially those who were started on successful sustained ART relatively early in the course of infection. This knowledge gap assumed additional importance in 2013 with the issuance of new American College of Cardiology/American Heart Association (ACC/AHA) guidelines for treatment of hyperlipidemia [75]. Cholesterol-lowering drugs may be considered the most powerful and widely applicable available tool for reducing risk of CVD events. The updated lipid treatment recommendations move away from the traditional approach of identifying patients who may benefit from HMG coenzyme A reductase inhibitors (“statins”) on the basis of their having elevated levels of low-density lipoprotein cholesterol (LDL-c). Instead, physicians are to base decisions about patients’ eligibility for cholesterol-lowering therapy based upon the patient’s predicted future CVD risk. For example for populations free of clinical CVD or diabetes who have LDL-c levels in the range of 70 to 189 mg/dL, clinicians are advised to first estimate each patient’s future CVD risk according to the Pooled Cohort Equation, a new risk prediction equation that predicts 10-year risk for a first atherosclerotic CVD event depending upon sex, age, race, total and HDL-cholesterol levels, systolic blood pressure and antihypertensive treatment, and smoking. Those who exceed a 7.5 % risk threshold are eligible for statin treatment regardless of LDL-c levels.
Thus, the new cholesterol treatment guidelines introduce a new CVD risk prediction formula (Pooled Cohort Equation), which, like others such as the Framingham Risk Score, are of uncertain accuracy in HIV-infected individuals. The predicted CVD risk, rather than LDL-c levels, has now become a primary driver of decisions about statin lipid treatment eligibility and intensity. The ACC/AHA report did not recommend a specific strategy for considering HIV infection or other non-traditional risk factors for CVD in decisions about the use of statins. However, long-term cohort study data in the general (non-HIV infected) population support the conclusion that other non-traditional CVD risk markers such as C-reactive protein and coronary artery calcium score may help to select patients who would benefit from statin therapy [76]. What is the role of clinical markers of HIV disease will remain uncertain until additional longitudinal CVD follow-up data become available in HIV-infected populations. Meanwhile, the ACTG REPRIEVE trial (A5332, Randomized Trial to Prevent Vascular Events in HIV) will for the first time address the efficacy of a HMG coenzyme A reductase inhibitor therapy (pitavastatin) versus placebo on major CVD events among 6500 subjects with chronic HIV infection over 72 months follow-up [77••].
Conclusions
As summarized above, new research continues to elucidate CVD in the HIV-infected population. While a large number of such studies are available, both the degree and the risk factors of CVD associated with HIV are likely to be different in the future as new strategies of earlier diagnosis and treatment of HIV infection are adopted in the community. Death due to CVD has decreased markedly over the past decade among the HIV-infected population. Paradoxically, however, CVD is now as important as ever as a cause of death because over the same period of time AIDS mortality has fallen even faster. As compared with their HIV-uninfected counterparts, the HIV-infected population as a whole has higher CVD event rates, even after matching and controlling for CVD risk factors that tend to be overrepresented in the HIV-infected population. Markers of poor HIV disease control including low CD4+ T cell count and high HIV RNA identify those individuals with the highest risk of incident CVD, but even well-controlled HAART-treated individuals may have elevated CVD risk. Finally, patients with advanced and poorly controlled HIV disease may also be vulnerable to cardiac fibrosis and steatosis as well as other non-traditional mechanisms that exacerbate the CVD risk presented by classic atherosclerotic risk factors.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Palella Jr FJ, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV outpatient study investigators. N Engl J Med. 1998;338(13):853–60.
Wada N, Jacobson LP, Cohen M, et al. Cause-specific life expectancies after 35 years of age for human immunodeficiency syndrome-infected and human immunodeficiency syndrome-negative individuals followed simultaneously in long-term cohort studies, 1984–2008. Am J Epidemiol. 2013;177(2):116–25.
Samji H, Cescon A, Hogg RS, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS One. 2013;8(12), e81355.
INSIGHT START Study Group. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015.
Volberding PA, Deeks SG. Antiretroviral therapy and management of HIV infection. Lancet. 2010;376(9734):49–62.
Mdodo R, Frazier EL, Dube SR, et al. Cigarette smoking prevalence among adults with HIV compared with the general adult population in the United States: cross-sectional surveys. Ann Intern Med. 2015;162(5):335–44.
Stein JH, Hsue PY. Inflammation, immune activation, and CVD risk in individuals with HIV infection. JAMA. 2012;308(4):405–6.
Shrestha S, Irvin MR, Grunfeld C, et al. HIV, inflammation, and calcium in atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34(2):244–50.
Stein JH, Currier JS, Hsue PY. Arterial disease in patients with human immunodeficiency virus infection what has imaging taught Us? J Am Coll Cardiol Img. 2014;7(5):515–25.
Ford ES, Capewell S. Proportion of the decline in cardiovascular mortality disease due to prevention versus treatment: public health versus clinical care. Annu Rev Public Health. 2011;32:5–22.
Ford ES, Ajani UA, Croft JB, et al. Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N Engl J Med. 2007;356(23):2388–98.
Helleberg M, Kronborg G, Larsen CS, et al. Causes of death among Danish HIV patients compared with population controls in the period 1995–2008. Infection. 2012;40(6):627–34.
Smith CJ, Ryom L, Weber R, et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet. 2014;384(9939):241–8. Among 49,731 HIV infected study participants seen at clinics in Europe, the U.S., and Australia, the age-standardized CVD mortality rate decreased from 2.0/1,000 person-years in 1999 to 0.7/1,000 person-years in 2011.
Hanna DB, Ramaswamy C, Kaplan RC, et al. Cardiovascular disease mortality among HIV-infected persons, New York City, 2001–2012 [abstract 729]. Seattle: Conference on Retroviruses and Opportunistic Infections; 2015.
Adih WK, Selik RM, Hu X. Trends in diseases reported on US death certificates that mentioned HIV infection, 1996–2006. J Int Assoc Phys AIDS Care. 2011;10(1):5–11.
Ingle SM, May MT, Gill MJ, et al. Impact of risk factors for specific causes of death in the first and subsequent years of antiretroviral therapy among HIV-infected patients. Clin Infect Dis. 2014;59(2):287–97.
Morlat P, Roussillon C, Henard S, et al. Causes of death among HIV-infected patients in France in 2010 (national survey): trends since 2000. AIDS. 2014;28(8):1181–91.
Schwarcz SK, Vu A, Hsu LC, et al. Changes in causes of death among persons with AIDS: San Francisco, California, 1996–2011. AIDS Patient Care STDS. 2014;28(10):517–23.
Rodger AJ, Lodwick R, Schechter M, et al. Mortality in well controlled HIV in the continuous antiretroviral therapy arms of the SMART and ESPRIT trials compared with the general population. AIDS. 2013;27(6):973–9. Adults with HIV infection who are well-controlled on ART have mortality rates close to that observed among the general population.
Lewden C, Bouteloup V, De Wit S, et al. All-cause mortality in treated HIV-infected adults with CD4 >/=500/mm3 compared with the general population: evidence from a large European observational cohort collaboration. Int J Epidemiol. 2012;41(2):433–45.
Freiberg MS, Chang CC, Kuller LH, et al. HIV infection and the risk of acute myocardial infarction. JAMA internal medicine. 2013;173(8):614–22. After adjusting for traditional CVD risk factors, comorbidities, and substance use, HIV-infected veterans had an increased risk of incident MI compared with uninfected veterans. This was a large population based study featuring medical records review to confirm incident myocardial infarction events.
Klein DB, Leyden WA, Xu LF, et al. Declining relative risk for myocardial infarction among HIV-positive compared with HIV-negative individuals with access to care. Clin Infect Dis. 2015;60(8):1278–80. In the Kaiser Permanente Northern California population, the adjusted relative risk (RR) of incident MI was 1.4 (95% CI, 1.2–1.6) comparing the HIV-infected group versus the HIV-uninfected group. The pattern of results over time suggested declining relative risk in the HIV-infected population as compared with HIV-uninfected comparators over time, which may reflect a high prevalence of statin use in the latter years of the study that might have diminished the HIV-related relative risk.
Silverberg MJ, Leyden WA, Xu LF, et al. Immunodeficiency and risk of myocardial infarction among HIV-positive individuals with access to care. J Acquir Immune Defic Syndr. 2014;65(2):160–6.
Crane HM, Heckbert SR, Drozd DR, et al. Lessons learned from the design and implementation of myocardial infarction adjudication tailored for HIV clinical cohorts. Am J Epidemiol. 2014;179(8):996–1005. Among HIV patients from eight U.S. clinical sites, approximately half of MI events were caused by atypical mechanisms such as vasospasm from use of cocaine or sepsis, which can precipitate an acute CVD event above and beyond traditional CVD risk factors.
Sabin CA, Ryom L, De Wit S, et al. Associations between immune depression and cardiovascular events in HIV infection. AIDS. 2013;27(17):2735–48.
Lang S, Mary-Krause M, Simon A, et al. HIV replication and immune status are independent predictors of the risk of myocardial infarction in HIV-infected individuals. Clin Infect Dis. 2012;55(4):600–7.
Triant VA, Regan S, Lee H, et al. Association of immunologic and virologic factors with myocardial infarction rates in a US healthcare system. J Acquir Immune Defic Syndr. 2010;55(5):615–9.
Hanna DB, Post WS, Deal JA, et al. HIV infection is associated with progression of subclinical carotid atherosclerosis. Clin Infect Dis. 2015;61(4):640–50. Among 199 HIV-infected participants who were receiving ART and persistently virologically suppressed over 7 years, this study observed an increased risk of new focal carotid artery plaque formation compared with the HIV-uninfected group (adjusted RR = 1.77, 95% CI, 1.13 – 2.77). While limited to a measure of subclinical atherosclerosis, this report is notable for documenting increased CVD risk among individuals maintained long-term on suppressive ART therapy.
Sani MU. Myocardial disease in human immunodeficiency virus (HIV) infection: a review. Wien Klin Wochenschr. 2008;120(3–4):77–87.
Thienemann F, Sliwa K, Rockstroh JK. HIV and the heart: the impact of antiretroviral therapy: a global perspective. Eur Heart J. 2013;34(46):3538–46.
Cerrato E, D'Ascenzo F, Biondi-Zoccai G, et al. Cardiac dysfunction in pauci symptomatic human immunodeficiency virus patients: a meta-analysis in the highly active antiretroviral therapy era. Eur Heart J. 2013;34(19):1432–6.
Redfield MM, Jacobsen SJ, Burnett Jr JC, et al. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA. 2003;289(2):194–202.
Samdarshi TE, Taylor HA, Edwards DQ, et al. Distribution and determinants of doppler-derived diastolic flow indices in African Americans: the Jackson heart study (JHS). Am Heart J. 2009;158(2):209–16.
Blecker S, Matsushita K, Fox E, et al. Left ventricular dysfunction as a risk factor for cardiovascular and noncardiovascular hospitalizations in African Americans. Am Heart J. 2010;160(3):488–95.
Brooks JT, Buchacz K, Gebo KA, et al. HIV infection and older Americans: the public health perspective. Am J Public Health. 2012;102(8):1516–26.
Cade WT. Left ventricular dysfunction in human immunodeficiency virus infection. J Cardiometab Syndr. 2008;3(2):83–7.
Mentz RJ, Kelly JP, von Lueder TG, et al. Noncardiac comorbidities in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol. 2014;64(21):2281–93.
Butt AA, Chang CC, Kuller L, et al. Risk of heart failure with human immunodeficiency virus in the absence of prior diagnosis of coronary heart disease. Arch Intern Med. 2011;171(8):737–43. A study of U.S. veterans showed HIV infection to be associated with an almost 2-fold risk of incident heart failure in the absence of previously documented coronary heart disease.
Hsu JC, Li Y, Marcus GM, Hsue PY, Scherzer R, Grunfeld C, et al. Atrial fibrillation and atrial flutter in human immunodeficiency virus-infected persons: incidence, risk factors, and association with markers of HIV disease severity. J Am Coll Cardiol. 2013;61(22):2288-95. doi: 10.1016/j.jacc.2013.03.022. Epub 2013 Apr 3.
Tseng ZH, Secemsky EA, Dowdy D, et al. Sudden cardiac death in patients with human immunodeficiency virus infection. J Am Coll Cardiol. 2012;59(21):1891–6.
Ho JE, Hsue PY. Cardiovascular manifestations of HIV infection. Heart. 2009;95(14):1193–202.
Hsue PY, Deeks SG, Hunt PW. Immunologic basis of cardiovascular disease in HIV-infected adults. J Infect Dis. 2012;205 Suppl 3:S375–82.
Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263–71.
Mosunjac MI, Sundstrom JB, Heninger M, et al. Combined pathological effects of cocaine abuse and HIV infection on the cardiovascular system: an autopsy study of 187 cases from the Fulton county medical Examiner's office. Am J Forensic Med Pathol. 2008;29(1):9–13.
Seltenhammer MH, Marchart K, Paula P, et al. Micromorphological changes in cardiac tissue of drug-related deaths with emphasis on chronic illicit opioid abuse. Addiction. 2013;108(7):1287–95.
Laonigro I, Correale M, Di Biase M, et al. Alcohol abuse and heart failure. Eur J Heart Fail. 2009;11(5):453–62.
Remick J, Georgiopoulou V, Marti C, et al. Heart failure in patients with human immunodeficiency virus infection: epidemiology, pathophysiology, treatment, and future research. Circulation. 2014;129(17):1781–9.
Group DADS, Sabin CA, Worm SW, et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet. 2008;371(9622):1417–26.
Choi AI, Vittinghoff E, Deeks SG, et al. Cardiovascular risks associated with abacavir and tenofovir exposure in HIV-infected persons. AIDS. 2011;25(10):1289–98.
Ding X, Andraca-Carrera E, Cooper C, et al. No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis. J Acquir Immune Defic Syndr. 2012;61(4):441–7.
Strategies for Management of Antiretroviral Therapy Study G, El-Sadr WM, Lundgren J, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355(22):2283–96.
Group DADS, Friis-Moller N, Reiss P, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med. 2007;356(17):1723–35.
Lang S, Mary-Krause M, Cotte L, et al. Impact of individual antiretroviral drugs on the risk of myocardial infarction in human immunodeficiency virus-infected patients: a case–control study nested within the French Hospital Database on HIV ANRS cohort CO4. Arch Intern Med. 2010;170(14):1228–38.
Kaplan RC, Kingsley LA, Sharrett AR, et al. Ten-year predicted coronary heart disease risk in HIV-infected men and women. Clin Infect Dis. 2007;45(8):1074–81.
Monforte A, Reiss P, Ryom L, et al. Atazanavir is not associated with an increased risk of cardio- or cerebrovascular disease events. AIDS. 2013;27(3):407–15.
Gazoti Debessa CR, Mesiano Maifrino LB, de Souza Rodrigues R. Age related changes of the collagen network of the human heart. Mech Ageing Dev. 2001;122(10):1049–58.
Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32(6):670–9.
Mewton N, Liu CY, Croisille P, et al. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. 2011;57(8):891–903.
Ambale-Venkatesh B, Lima JA. Cardiac MRI: a central prognostic tool in myocardial fibrosis. Nat Rev Cardiol. 2015;12(1):18–29.
Liu CY, Liu YC, Wu C, et al. Evaluation of age-related interstitial myocardial fibrosis with cardiac magnetic resonance contrast-enhanced T1 mapping: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol. 2013;62(14):1280–7.
Ambale Venkatesh B, Volpe GJ, Donekal S, et al. Association of longitudinal changes in left ventricular structure and function with myocardial fibrosis: the multi-ethnic study of atherosclerosis study. Hypertension. 2014;64(3):508–15.
Lopaschuk GD, Ussher JR, Folmes CD, et al. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–58.
Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15(6):805–12.
Rajabi M, Kassiotis C, Razeghi P, et al. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. 2007;12(3–4):331–43.
McGavock JM, Lingvay I, Zib I, et al. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. 2007;116(10):1170–5.
Utz W, Engeli S, Haufe S, et al. Myocardial steatosis, cardiac remodelling and fitness in insulin-sensitive and insulin-resistant obese women. Heart. 2011;97(19):1585–9.
van der Meer RW, Rijzewijk LJ, Diamant M, et al. The ageing male heart: myocardial triglyceride content as independent predictor of diastolic function. Eur Heart J. 2008;29(12):1516–22.
Mahmod M, Bull S, Suttie JJ, et al. Myocardial steatosis and left ventricular contractile dysfunction in patients with severe aortic stenosis. Circ Cardiovasc Imaging. 2013;6(5):808–16.
Rijzewijk LJ, van der Meer RW, Smit JW, et al. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. 2008;52(22):1793–9.
Ng AC, Delgado V, Bertini M, et al. Myocardial steatosis and biventricular strain and strain rate imaging in patients with type 2 diabetes mellitus. Circulation. 2010;122(24):2538–44.
Korosoglou G, Humpert PM, Ahrens J, et al. Left ventricular diastolic function in type 2 diabetes mellitus is associated with myocardial triglyceride content but not with impaired myocardial perfusion reserve. J Magn Reson Imaging. 2012;35(4):804–11.
Holloway CJ, Ntusi N, Suttie J, et al. Comprehensive cardiac magnetic resonance imaging and spectroscopy reveal a high burden of myocardial disease in HIV patients. Circulation. 2013;128(8):814–22. A cardiac magnetic resonance imaging study in HIV infected patients receiving combination ART found increased left ventricular patchy fibrosis by late gadolinium enhancement and fat content by MRS, along with depressed measures of myocardial deformation.
Thiara DK, Liu CY, Raman F, et al. Abnormal myocardial function is related to myocardial steatosis and diffuse myocardial fibrosis in HIV-infected adults. J Infect Dis. 2015;212(10):1544–51.
Nelson MD, Szczepaniak LS, LaBounty TM, et al. Cardiac steatosis and left ventricular dysfunction in HIV-infected patients treated with highly active antiretroviral therapy. J Am Coll Cardiol Img. 2014;7(11):1175–7.
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults a report of the American College of Cardiology/American Heart Association task force on practice guidelines. Circulation. 2014;129(25):S1–S45.
Yeboah J, Polonsky TS, Young R, et al. Utility of nontraditional risk markers in individuals ineligible for statin therapy according to the 2013 American College of Cardiology/American Heart Association cholesterol guidelines. Circulation. 2015;132(10):916–22.
Mitka M. Exploring statins to decrease HIV-related heart disease risk. J Am Med Assoc. 2015;314(7):657–9. The ACTG REPRIEVE trial (A5332, Randomized Trial to Prevent Vascular Events in HIV) will for the first time address the efficacy of a HMG coenzyme A reductase inhibitor therapy (pitavastatin) versus placebo on major CVD events among 6500 subjects with chronic HIV infection over 72 months follow-up.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Robert C. Kaplan, David B. Hanna, and Jorge R. Kizer declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on HIV Pathogenesis and Treatment
Rights and permissions
About this article

Cite this article
Kaplan, R.C., Hanna, D.B. & Kizer, J.R. Recent Insights Into Cardiovascular Disease (CVD) Risk Among HIV-Infected Adults. Curr HIV/AIDS Rep 13, 44–52 (2016). https://doi.org/10.1007/s11904-016-0301-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11904-016-0301-4