
Abstract
Chronic myelomonocytic leukemia is a clonal malignancy of the ageing hematopoietic stem cell characterized by a biased differentiation leading to persistent monocytosis and inconstant hypersensitivity of myeloid progenitors to granulo-monocyte colony-stimulating factor (GM-CSF). Cytogenetic abnormalities identified in 30–40 % of patients and gene mutations detected in every patient can be used to stratify patients into risk groups that guide the therapeutic choices. TET2, SRSF2, ASXL1, and genes of the Ras pathway are the most frequently mutated genes, with ASXL1 mutations negatively affecting the disease outcome. Allogeneic stem cell transplantation is the first option to consider, especially in younger patients with poor prognostic factors. There is no firm clinical guideline in transplant-ineligible patients, but hypomethylating agents might be an interesting option. A consensus prognostic scoring system and specific response criteria are now required to facilitate the evaluation of new therapeutic strategies in clinical trials specifically dedicated to this disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic myelomonocytic leukemia (CMML) is a clonal hematopoietic malignancy of the elderly with an estimated incidence of 1 case per 100,000 inhabitants per year and a clear male predominance (sex ratio = 2). The median age at diagnosis is ~70 and the disease is exceptionally diagnosed before 50 years of age [1, 2]. CMML associates features of both myeloproliferative neoplasia (MPN) and myelodysplastic syndrome (MDS), which have long tantalized biologists and clinicians attempting to classify and to treat this disease.
In 1982, the French-American-British (FAB) classification incorporated CMML as a subgroup in MDS because of dysplastic changes commonly found in CMML blood and bone marrow cells, with cytopenias and potential risk of transformation into acute myeloid leukemia (AML) [3, 4]. However, a fraction of CMML patients have high white blood cell (WBC) counts, with sometimes a splenomegaly, skin infiltration, and serous effusions [5, 6], leading the FAB group to define two subsets of CMML in 1994, namely, a dysplastic type (MD-CMML), and a proliferative type (MP-CMML), using a cut point of WBC at 13 G/L [7].
Seven years later, the world health organization (WHO) individualized a new class of hematological malignancies with both myeloproliferative and myelodysplastic characteristics, CMML being the most frequent of them [8, 9]. The WHO committee also defined two subgroups, based on blast cell percentage in the blood and the bone marrow, namely, CMML-1 (<5 % in the blood, <10 % in the bone marrow) and CMML-2 (5–19 % in the blood, 10–19 % in the bone marrow, or less if Auer inclusions are present).
Finally, in 2008, a novel designation was given to these overlap diseases, the myelodysplasic syndromes/myeloproliferative neoplasia (MDS/MPN), that include CMML as the most frequent entity [10•], comprising more than 90 % of all MDS/MPN cases [2], the juvenile myelomonocytic leukemia (JMML) a pediatric disease with clinical syndrome similar to that of CMML [11], the atypical chronic myeloid leukemia (a-CML), and the unclassifiable MDS/MPNs (MDS/MPN-U) [12••]. The WHO classification added to these diseases refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) as a provisional entity [13] (Table 1).
Very few clinical trials have been specifically dedicated to CMML in the last decades, patients with an aggressive CMML being usually included as a subgroup of patients in trials dedicated to high-grade MDS [14]. However, in the last 5 years, we have been facing an unprecedented flurry of information that dissects the distinctiveness of CMML. We are coming closer to understanding the pathogenesis of the disease, although major questions and challenges are still to be addressed. Here, we discuss how the recent insights in the pathogenesis of CMML influence our current practice and might impact the evaluation of new therapeutic approaches in trials dedicated to this disease.
CMML Diagnosis
CMML is defined by the WHO as a clonal disease of the bone marrow hematopoietic stem cell characterized by persistent monocytosis. The clinical features of the disease are nonspecific and highly variable from a patient to another. In most cases, the onset of the disease is insidious and the diagnosis is incidental when a complete blood count is obtained for unrelated reasons. Clinical symptoms related to cytopenias (anemia, infections, bleeding) can be inaugural, and patients with a proliferative form of the disease can demonstrate weight loss, drenching night sweats, abdominal discomfort, and splenomegaly [15]. Occasionally, skin infiltration with abnormal monocytes (leukemia cutis) is the initial manifestation [16–18]. Some patients may directly present in the blastic phase of CMML as acute myeloid leukemia (AML). Similar to therapy-related MDS, CMML following cytotoxic chemotherapy has also been reported [19].
The diagnosis of CMML is currently based on the criteria proposed by the WHO in 2008 [10•], which include a unique positive criterion: monocyte count in the peripheral blood must be higher than 1 g/L for at least 3 months in the absence of other cause of monocytosis. The diagnosis is enforced when the cytologists identify dysplastic features in one or several myeloid lineage but, given the subjectivity of bone marrow cell dysplasia, this aspect is not an absolute requirement for CMML diagnosis.
The WHO classification also recommends to exclude (1) an AML defined by a percentage of blast cells (including myeloblasts, monoblasts, and promonocytes) in the bone marrow higher than 20 %, (2) a chronic myeloid leukemia with monocytosis identified by the presence of a Philadelphia chromosome or a BCR-ABL fusion gene in bone marrow cells, and (3) a disease entering the so-called Myeloid and lymphoid neoplasm with eosinophilia (MLN-eo) category in which a gene rearrangement involves the platelet-derived growth factor receptor (PDGFR-A or PDGFR-B) gene and confers to leukemic cells a sensitivity to some tyrosine kinase inhibitors such as imatinib [20, 21].
Since monocytosis is the unique positive criterion in the disease definition by the WHO, it is of prime importance to exclude a secondary monocytosis [22, 23]. The main causes of reactive monocytosis are (1) infectious diseases such as tuberculosis, brucellosis, bacterial endocarditis, fungal infections, protozoan infections, and chronic viral infections; (2) chronic inflammatory diseases including autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus, sarcoidosis, lipid storage familial disorders, and metastatic cancers; and (3) bone marrow regeneration after therapeutic and other aplasias, which usually does not meet the 3 month duration criterion.
The persistence of the monocytosis during 3 months delays the moment when the diagnostic is made. In addition, the disease presentation is heterogeneous and CMML diagnosis can be difficult in several circumstances. First, CMML patients demonstrate an abnormally high rate of autoimmune diseases as compared to age-matched population, sometimes making the distinction between CMML and reactive monocytosis confused [24, 25]. Second, some patients with a primitive myelofibrosis (PMF) in proliferative phase with considerable leucocytosis meet the WHO criteria for CMML, whereas other patients with a low WBC (1 g/L) and a high percentage of monocytes do not [26]. Due to these diagnostic difficulties and the lack of therapeutic consequences of CMML assimilation to MDS so far, the disease frequency could have been underestimated. The number of MDS that secondary develop a monocytosis, then fulfill the CMML diagnosis criteria, is also poorly known as well as the specificities, if any, of the rare cases that appear below 60 years.
Additional criteria are needed to improve CMML identification. The presence of acquired cytogenetic and genetic abnormalities in bone marrow and blood cells supports CMML diagnosis in ambiguous situations, e.g., in patients with an isolated monocytosis without any dysplastic feature and in those who show auto-immune disorders with an inflammatory reaction. Although none of these cytogenetic and genetic abnormalities is specific of the disease, their detection identifies clonal hematopoiesis [27]. Other criteria, including the phenotype of peripheral blood monocytes, are currently tested as biological markers that could facilitate the distinction between primitive and reactive monocytosis, suppress the 3-month delay to diagnosis, and revisit the definition of the cutoff value of monocytosis, currently at 1 G/L (DSB, ES, unpublished data).
Cellular Features
CMML initiating cells remain a disputed issue as long-term cultures and serial replating assays have led to distinct conclusion and mouse xenotransplantation assays have shown limited engraftment [28]. Ongoing effort using new mouse strains and more selective cell sorting might allow a more precise identification of initiating cells.
An important feature of JMML is the hypersensitivity of myeloid progenitors to granulo-monocyte colony-stimulating factor (GM-CSF), as measured by hematopoietic colony formation and GM-CSF-dependent phosphorylation of STAT5 [29]. This hypersensitivity is a more controversial issue in CMML, in which we identified two subgroups, depending on the presence or absence of a constitutive activation of signaling pathways [30••]. However, others identified an elevated proportion of immature GM-CSF receptor-α (R) subunit-expressing cells in the bone marrow of CMML patients and a response of myeloid and monocytic progenitors to GM-CSF signal inhibition [31••]. The bone marrow microenvironment could also affect disease evolution, e.g., through a decreased production of IL-32 [32].
Among the more differentiated cells of the clone, we have noticed that the cells recognized by cytologists as monocytes in the peripheral blood include a variable proportion of immature dysplastic granulocytes. We have shown also that these cells, which belong to the leukemic clone as they demonstrate the same genetic alterations as compared to monocytes, synthesize and secrete large quantities of the antimicrobial peptides alpha-defensins that inhibit M-CSF-induced differentiation of monocytes into macrophages through the purinergic receptor P2Y7 [33••]. Since these dysplastic and immature granulocytes also kill activated T cells, they are reminiscent of the myeloid-derived suppressive cells described in patients with MDS or metastatic solid tumor.
Genetic Abnormalities
Conventional cytogenetic analysis of bone marrow cells detects recurrent abnormalities in 30–40 % of CMML cases. None of these aberrations that include aneuploidies such as trisomy 8, monosomy 7, and interstitial deletions of chromosomes 20q, 12p, 7q, and 13q as the most frequent is specific of the disease as they are common to a range of myeloid disorders [27]. A minority of patients present with reciprocal translocations leading to fusions involving diverse tyrosine kinase genes [34, 35]. Some fusion genes, such as those involving ABL1, confer sensitivity to imatinib whereas others such as those involving JAK2 or FGFR1 rather confer sensitivity to ruxolitinib or ponatinib, respectively, but these fusions genes are very rare in CMML [36–38]. High-resolution single-nucleotide polymorphism arrays (SNP-A) and comparative genomic hybridization (CGH) detect copy number abnormalities and loss of heterozygosity due to uniparental disomy (copy neutral loss of heterozygosity) in about 50 % of patients [39]. In addition, to enforce the diagnosis of ambiguous monocytosis, these abnormalities define evolutionary risk subgroups.
Whole exome sequencing of peripheral blood sequencing detects more than 10 gene mutations in every patient [40••] (Merlevede J, DSB, ES unpublished data). Again, none of the recurrent mutations identified in patient monocytes is disease specific. A broad pattern of recurrent mutations, affecting ~40 distinct genes, has been detected. The most frequent mutations affect TET2 (up to 60 % of patients in some series, CMML is the most frequent neoplasm with TET2 gene mutation), SRSF2 (up to 50 % of cases), and ASXL1 (about 40 % of cases) [41••].
The recurrent mutations identified in CMML can be grouped as those that impact the (1) epigenetic control of transcription through regulating DNA methylation (DNMT3A) and demethylation (TET2, IDH1, IDH2), histone modification (ASXL1, EZH2, UTX), and nuclear cohesin complex components (STAG2, SMC3, SMC1A, and RAD21); (2) pre-mRNA splicing (SRSF2, SF3B1, U2AF1, ZRSR2); (3) cell signaling (KRAS, NRAS, CBL, JAK2, CSF3R, and genes of the Notch2 pathway); and (4) transcription factors such as RUNX1 and, in rare cases, TP53 or NPM1. In a given subgroup, gene mutations are usually mutually exclusive. Detailed reviews of the functional consequences of these diverse gene mutations have been published recently [42]. Mutations leading to the constitutive activation of signaling pathways correlate with a myeloproliferative phenotype and an enhanced sensitivity of myeloid progenitors to GM-CSF [30••].
Analysis of the CMML clonal architecture by mutation-specific discrimination analysis at the level of single-cell-derived colonies suggested early clonal dominance with the linear accumulation of mutations in the stem cell compartment (CD34+, CD38−, CD90+), a few branching events due to mitotic recombination events that are death routes in clonal evolution, and a clonal sweep of the most mutated cells with differentiation (Fig. 1). The first mutation to appear often affects an epigenetic gene, especially TET2 or ASXL1, preceding the mutation of a splicing factor gene, most often SRSF2, which, in the proliferative forms of the disease, is followed by a mutation in a signaling gene, leading in many case to the constitutive activation of the Ras pathway. However, alternative orders of appearance of gene mutations do exist, without clearly identified relationship between this order of appearance of mutations and the disease phenotype. We have shown that the early clonal dominance of TET2 mutations could account for the bias of differentiation leading to overproduction of monocytes [30••].

Schematic representation of the clonal architecture in CMML, showing clonal dominance (very few wild-type cells in the hematopoietic stem cell compartment in which the mutations accumulate linearly) and clonal sweep of the most mutated cells with differentiation [30••]
Epigenetic alterations of gene expression do exist also in CMML cells. For example, hypermethylation of TIF1γ gene promoter leading to gene expression downregulation was identified in 35 % of CMML patients and the demethylation of this gene promoter, together with gene re-expression, could be used as a biomarker of hypomethylating agent activity, although this methylation pattern does not predict the drug efficacy [43••, 44]. When the current therapeutics used to treat CMML induce a clinical and biological response, they modify the clonal architecture by reducing the fitness of the most mutated cells in the malignant clone without eradicating these cells that reappear when the disease escapes to therapy [30••].
Several unsupervised analyses of gene expression in CMML cells have been reported. Such an analysis in bone marrow mononuclear cells discriminates two groups, based on the expression of genes that regulate the Jun kinase and NF-B pathways. In sorted peripheral blood CD14+ monocytes, we separated a minor subgroup of patients with a gene signature similar to that of normal monocytes, whereas most patients demonstrated an aberrant gene expression pattern in which overexpressed CJUN and CMYB were poor prognostic markers [44]. A prognostic signature was also validated in peripheral blood mononuclear cells, mostly composed of genes expressed in immature granulocytes [45].
Prognostic Criteria and Scores in CMML
The clinical course of CMML is highly variable, with some patients remaining stable for many years without any treatment while other rapidly progress. Therefore, life expectancy ranges from several months to several years. The mean overall survival is currently between 2.5 and 3 years, which is higher than MPS-PMN-U (mean survival ~2 years) and a-CML (mean survival ~1 year) among MDS/MPNs [12••].
Transformation into AML, which occurs in ~30 % of patients, is a common cause of death as these secondary AMLs are usually refractory to current therapeutic approaches [42]. Accordingly, the percentage of blast cells in the bone marrow and the blood is the only prognostic parameter recognized by the WHO classification [10•].
Other patients die without overt clinical disease progression. The consequences of cytopenias combine with age-associated co-morbidities and progressive increase in disease-related symptom burden, leading to early death. These symptoms and their impact on quality of life have not been specifically explored in CMML as they are in other diseases [46, 47]. As MDS patients, CMML patients suffer from debilitating fatigue, impaired mobility, suboptimal self-care, night sweats, bone pain, discomfort, fever, skin rash, and weight loss.
In order to anticipate the disease evolution and to propose the best therapeutic program, risk stratification is critical to establish. The international prognostic scoring system (IPSS) for survival stratified MDS patients into four risk groups (low, intermediate 1, intermediate 2, and high) [48], based on a cohort of patients that included CMML. However, neither the IPSS score nor its revised version [49] could apply to MP-CMML with leucocytosis greater than 12 G/L.
Several prognostic scores based on routinely available clinical and biological variables have been established by groups in France (Lille 1988 and 1993) [50, 51], England (Bournemouth 1988) [52], Spain (1989) [53], Germany (Düsseldorf, 1992) [54], and USA (MD Anderson, 2002) [55]. These analyses identified splenomegaly, hemoglobin level, platelet, lymphocyte or monocyte counts, and medullary blast cell percentage as prognostic factors in univariate analysis, whereas bone marrow blast count and hemoglobin level remained usually significant in multivariate analysis (for detailed comparison of these prognostic scores, see [56]). However, with the exception of the modified Bournemouth score (based on FAB criteria and excluding CMML with more than 5 % blast cells in the blood), these prognostic scores are biased toward MDS as they were not specific of the disease.
A CMML-specific prognostic score based on routine biological parameters was established by the Mayo Clinic [57] and combined an increased peripheral monocyte count with leukocytosis, anemia, and thrombocytopenia to classify CMML patients into three groups with an overall median survival of 32.0, 18.5, and 10.0 months in the low, intermediate, and high-risk groups, respectively.
In 2013, a Spanish group highlighted the prognostic role cytogenetic anomalies by introducing the karyotype in a new, CMML-specific, prognostic score (CPSS for CMML-specific prognostic scoring system) [58] in which the most relevant prognostic factors were the FAB and WHO classification, the red blood cell transfusion dependency, and the cytogenetic abnormalities (Table 2). This score ranked patients in four risk groups, including low, intermediate 1, intermediate 2, and high risk, with a median overall survival of 72, 31, 13, and 5 months, respectively. In these groups, the probability of AML transformation at 5 years was 13, 29, 60, and 73 %, respectively.
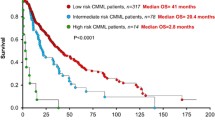
In 2013 also, gene mutations were introduced into CMML scoring systems. Independent associations of genetic variations in ASXL1, RUNX1, NRAS, CBL, TET2, EZH2 [59], SRSF2 [60], SETBP1 [61], and CSF3R [62] have been associated individually with disease outcome. By sequencing 19 candidate genes (TET2, ASXL1, EZH2, IDH1, IDH2, DNMT3A, SF3B1, SRSF2, ZRSF2, U2AF1, RUNX1, NPM1, TP53, NRAS, KRAS, CBL, JAK2, FLT3, and KIT) in a cohort of 312 patients [41••], we have shown that mutations in ASXL1 gene were the only gene alterations that predict overall survival and AML transformation in multivariate analysis (Table 2). Other significant parameters were age (older than 65 years), WBC, platelet count, and hemoglobin level. We proposed a new prognosis scoring system that merges these parameters to rank patients into three groups of significant size (none including less than 20 % of patients) with low risk (overall survival not reached), intermediate (overall survival 380.5 months), and high (overall survival 140.4 months) risk. This score was validated on an independent cohort of 165 patients, and its validity was further confirmed recently in the Mayo Clinic cohort of patients [63].
An international working group is now needed to merge and validate existing databases, then definitely establish the usefulness of detecting ASXL1 mutations, and possibly other molecular aberrations, to stratify patients into risk groups that will guide the therapeutic choices. Such a validation will lead to the standardization of new routine laboratory assays. The total number of gene mutations in peripheral blood monocytes is another discriminating prognostic factor [30••], but logistic issues still preclude the implementation of whole exome/whole genome sequencing—or sequencing of a large panel of candidate genes—in routine clinical practice.
CMML Treatment
Allogeneic stem cell transplantation is the first option to consider in CMML patients, as it is the only therapy that was demonstrated to alter the natural history of the disease and to potentially cure some patients [22, 23]. Unfortunately, this treatment is rarely feasible, due to the advanced age of most patients. This is one of the reasons why there have been no randomized trial to precisely explore its impact in CMML patients. Retrospective studies indicate that overall survival ranges from 30 to 40 % [64–67]. Baseline factors that affect transplantation efficacy remain controversial. Although variable among series, relapse rate and treatment-related mortality appear to be high in CMML patients as compared to those with other diseases. The use of peripheral blood stem cells and transplants from matched unrelated donors [65], the advent of reduced intensity conditioning [68] and new options for preventing or treating graft versus host disease [69], may contribute to increase the number of allografted patients and improve their survival.
In transplant-ineligible CMML patients, there is no firm clinical guideline and very few clinical studies focused on this disease. When the disease was individualized in the 1980s, treatment options were confined to a “wait and watch” approach and best supportive care. These options are still considered in patients with nonproliferative, noncytopenic CMML-1.
In 1996, a randomized trial dedicated to poor-risk CMML patients demonstrated better overall survival in CMML patients treated with 1,000 mg/day of oral hydroxyurea compared to 150 mg/week of oral etoposide [70]. In the following years, hydroxyurea was largely used when the disease became proliferative. Meanwhile, several nonrandomized trials evaluated cytarabine [71], all-trans retinoic acid [72, 73], farnesyltransferase inhibitors [74], topoisomerase I inhibitors [75], and histone deacetylase inhibitors [76], with limited response rates and significant toxicities.
The Food and Drug Administration has then approved two hypomethylating agents, azacitidine (AZA) and decitabine for the treatment of patients with CMML. In Europe, AZA only has been approved for CMML patients with WBC < 13 G/L and bone marrow blasts between 10 and 29 %. If the pivotal randomized studies that demonstrated the efficacy and safety of demethylating agents in MDS patients included a very small number of CMML cases [14], several phase II trials testing hypomethylating agents in CMML patients only have now been completed [77]. Altogether, response rates range from 30 to 40 % and median overall survivals range from 1 to 3 years, depending on the patient characteristics at baseline [78].
Multivariable analyses of several series suggested that bone marrow blast percentage and WBC or monocyte counts at the start of AZA therapy were the parameters that impacted the overall survival rate [79]. In a prospective trial testing decitabine in 39 patients with aggressive CMML, we observed that overexpression of two known oncogenes, CJUN and CMYB, could predict inferior response rate and survival, respectively. On the other hand, neither gene mutations nor hypermethylation of the TIF1γ gene promoter was predictive of response or survival [44], and long-term analysis of this cohort suggested that decitabine could erase the negative prognostic impact of ASXL1 mutations in CMML patients [80].
Nevertheless, the benefit of hypomethylating agents in CMML patients remains controversial. Therefore, a randomized phase III trial testing decitabine versus hydroxyurea has been launched in Europe. CMML patients are included if they fulfill at least two severity criteria (marrow blasts higher than 5 %, clonal cytogenetic abnormality, hemoglobin lower than 100 g/L, absolute neutrophil count (ANC) higher than 16 G/L in the absence of infection, platelet count lower than 100 G/L, and splenomegaly) or if they show a documented extramedullary lesion, including cutaneous infiltration and pleural or pericardial effusion.
Emerging therapy include agents that may improve cytopenias such as thrombopoietin receptor agonists as eltrombopag [81] or romiplostim [82, 83], used to promote platelet production, or sotatercept, a chimeric protein containing the extracellular domain of the activin receptor 2A fused to the Fc domain of the human IgG1, used to promote erythropoiesis [84]. There are some concerns with the use of the thrombopoietin receptor agonist romiplostim about the risk of promoting acute leukemia transformation and marrow fibrosis, although this risk has still to be confirmed [83, 85]. Novel agents such as oral clofarabine (nucleoside analog) and lenalidomide (immunomodulatory agent) are also investigated.
New therapies could also intend to address GM-CSF hypersensitivity of CD38 expressing bone marrow myeloid progenitor population, which is a characteristic feature of CMML [30••, 31••]. In survival assays, myeloid progenitors are sensitive to GM-CSF signal inhibition with KB003, an anti-GM-CSF monoclonal antibody [30••]. However, the development strategy of this antibody, which has been disappointing in the treatment of severe asthma, is unknown. Based on the fact that GM-CSF receptor signals through JAK2 and that JAK2V617F mutation is identified in ~10 % of CMML patients, the safety and efficacy of JAK2 inhibition with ruxolitinib is currently evaluated in a phase II trial (moffitt.org/research--clinical-trials). The use of other JAK2 inhibitor with distinct spectrum of activity and the combination of these molecules with a demethylating agent could be explored in the future. Other putative targets include small-molecule inhibitors directed against STAT3/5, p38 mitogen- activating protein kinases (MAPK), AKT, and P13K-mTORC pathways.
Conclusion
Recent pathological investigations have strongly enforced the distinctiveness of CMML among other myeloid malignancies. Currently, disease treatment has to be considered in high-risk patients, based on refined prognostic scores including cytogenetic or molecular information. Allogeneic stem cell transplantation must be considered first in younger patients with high-risk CMML, whereas older patients with co-morbidity will be best suited for clinical trials comparing demethylating agents to hydroxyurea or testing novel agents. Major questions and challenges are still to be addressed, including the existence of pre-leukemic steps and the role of epigenetic, bone marrow microenvironment, and immune response escape in disease emergence and progression. The establishment of a consensus international prognostic scoring system that includes molecular alteration, and the definition of specific response criteria, might facilitate the evaluation of new therapeutic strategies in clinical trials specifically dedicated to the disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of outstanding importance
Phekoo KJ, Richards MA, Møller H, Schey SA, South Thames Haematology Specialist Committee. “The incidence and outcome of myeloid malignancies in 2,112 adult patients in southeast England,”. Haematologica. 2006;91(10):1400–4.
Osca-Gelis G, Puig-Vives M, Saez M, Gallardo D, Lloveras N, Marcos-Gragera R. Population-based incidence of myeloid malignancies: fifteen years of epidemiological data in the province of Girona, Spain. Haematologica. 2013;98(8):e95–97.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33(4):451–8.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51(2):189–99.
Michaux JL, Martiat P. Chronic myelomonocytic leukaemia (CMML)—a myelodysplastic or myeloproliferative syndrome? Leuk Lymphoma. 1993;9(1–2):35–41.
Germing U, Gattermann N, Minning H, Heyll A, Aul C. Problems in the classification of CMML—dysplastic versus proliferative type. Leuk Res. 1998;22(10):871–8.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick H, et al. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia. proposals by the French-American-British cooperative leukaemia group. Br J Haematol. 1994;87(4):746–54.
Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, et al. World health organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the clinical advisory committee meeting, Airlie house, Virginia, November 1997. J Clin Oncol Off J Am Soc Clin Oncol. 1999;17(12):3835–49.
Bennett JM. World health organization classification of the acute leukemias and myelodysplastic syndrome. Int J Hematol. 2000;72(2):131–3.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. “The 2008 revision of the world health organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes,”. Blood. 2009;114(5):937–51. The diagnosis criteria of CMML defined in 2008 by the WHO remain the reference, e.g. for including patients in dedicated clinical trials.
Loh ML. Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol. 2011;152(6):677–87.
Wang SA, Hasserjian RP, Fox PS, Rogers HJ, Geyer JT, Chabot-Richards D, et al. “Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms,”. Blood. 2014;123(17):2645–51. A recent retrospective analysis that clarifies the diagnosis criteria of 2 rare entities, atypical chronic myeloid leukemia and unclassifiable myelodysplastic/myeloproliferative neoplasm.
Malcovati L, Cazzola M. Refractory anemia with ring sideroblasts. Best Pract Res Clin Haematol. 2013;26(4):377–85.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–32.
Padron E, Komrokji R, List AF. The clinical management of chronic myelomonocytic leukemia. Clin Adv Hematol Oncol HO. 2014;12(3):172–8.
Manna M, Rehman HU. Medical image. Leukaemia cutis in myelomonocytic leukaemia. N Z Med J. 2012;125(1359):69–70.
Vitte F, Fabiani B, Bénet C, Dalac S, Balme B, Delattre C, et al. Specific skin lesions in chronic myelomonocytic leukemia: a spectrum of myelomonocytic and dendritic cell proliferations: a study of 42 cases. Am J Surg Pathol. 2012;36(9):1302–16.
Mathew RA, Bennett JM, Liu JJ, Komrokji RS, Lancet JE, Naghashpour M, et al. Cutaneous manifestations in CMML: indication of disease acceleration or transformation to AML and review of the literature. Leuk Res. 2012;36(1):72–80.
Takahashi K, Pemmaraju N, Strati P, Nogueras-Gonzalez G, Ning J, Bueso-Ramos C, et al. “Clinical characteristics and outcomes of therapy-related chronic myelomonocytic leukemia,”. Blood. 2013;122(16):2807–11.
Cheah CY, Burbury K, Apperley JF, Huguet F, Pitini V, Gardembas M, et al. Patients with myeloid malignancies bearing PDGFRB fusion genes achieve durable long-term remissions with imatinib. Blood. 2014;123(23):3574–7.
Gotlib J. World health organization-defined eosinophilic disorders: 2011 update on diagnosis, risk stratification, and management. Am J Hematol. 2011;86(8):677–88.
Onida F, Barosi G, Leone G, Malcovati L, Morra E, Santini V, et al. Management recommendations for chronic myelomonocytic leukemia: consensus statements from the SIE, SIES, GITMO groups. Haematologica. 2013;98(9):1344–52.
Parikh SA, Tefferi A. Chronic myelomonocytic leukemia: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88(11):967–74.
Braun T, Fenaux P. Myelodysplastic syndromes (MDS) and autoimmune disorders (AD): cause or consequence? Best Pract Res Clin Haematol. 2013;26(4):327–36.
J. Hadjadj, M. Michel, M.-P. Chauveheid, B. Godeau, T. Papo, and K. Sacre, “Immune thrombocytopenia in chronic myelomonocytic leukemia,” Eur. J. Haematol., Jun. 2014
Elliott MA, Verstovsek S, Dingli D, Schwager SM, Mesa RA, Li CY, et al. Monocytosis is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk Res. 2007;31(11):1503–9.
Such E, Cervera J, Costa D, Solé F, Vallespí T, Luño E, et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica. 2011;96(3):375–83.
Ramshaw HS, Bardy PG, Lee MA, Lopez AF. Chronic myelomonocytic leukemia requires granulocyte-macrophage colony-stimulating factor for growth in vitro and in vivo. Exp Hematol. 2002;30(10):1124–31.
Kotecha N, Flores NJ, Irish JM, Simonds EF, Sakai DS, Archambeault S, et al. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell. 2008;14(4):335–43.
Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, et al. “Clonal architecture of chronic myelomonocytic leukemias,”. Blood. 2013;121(12):2186–98. A study that describes the ordering of mutations in the leukemic clones and provides new pathogenic insights in CMML.
Padron E, Painter JS, Kunigal S, Mailloux AW, McGraw K, McDaniel JM, et al. “GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia,”. Blood. 2013;121(25):5068–77. Targeting hypersensitivity of bone marrow myeloid progenitors to GM-CSF is demonstrated as a potential therapeutic strategy.
Marcondes AM, Mhyre AJ, Stirewalt DL, Kim S-H, Dinarello CA, Deeg HJ. Dysregulation of IL-32 in myelodysplastic syndrome and chronic myelomonocytic leukemia modulates apoptosis and im pairs NK function. Proc Natl Acad Sci U S A. 2008;105(8):2865–70.
Droin N, Jacquel A, Hendra J-B, Racoeur C, Truntzer C, Pecqueur D, et al. “Alpha-defensins secreted by dysplastic granulocytes inhibit the differentiation of monocytes in chronic myelomonocytic leukemia,”. Blood. 2010;115(1):78–88. Evidence that circulating cells identified as monocytes include a variable proportion of dysplastic granulocytes endowed with immunosuppressive properties.
Costa D, Muñoz C, Carrió A, Nomdedeu M, Calvo X, Solé F, et al. “Reciprocal translocations in myelodysplastic syndromes and chronic myelomonocytic leukemias: review of 5,654 patients with an evaluable karyotype,”. Genes Chromosome Cancer. 2013;52(8):753–63.
Ballerini P, Struski S, Cresson C, Prade N, Toujani S, Deswarte C, et al. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia. 2012;26(11):2384–9.
B. J. Bain and S. Ahmad, “Should myeloid and lymphoid neoplasms with PCM1-JAK2 and other rearrangements of JAK2 be recognized as specific entities?,” Br. J. Haematol., Jun. 2014
Vardiman J, Hyjek E. World health organization classification, evaluation, and genetics of the myeloproliferative neoplasm variants. Hematol EducProgram Am Soc Hematol Am Soc Hematol Educ Program. 2011;2011:250–6.
Mozziconacci M-J, Carbuccia N, Prebet T, Charbonnier A, Murati A, Vey N, et al. Common features of myeloproliferative disorders with t (8;9) (p12;q33) and CEP110-FGFR1 fusion: report of a new case and review of the literature. Leuk Res. 2008;32(8):1304–8.
Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H, et al. 250 K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68(24):10349–57.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. “Frequent pathway mutations of splicing machinery in myelodysplasia,”. Nature. 2011;478(7367):64–9. The first description of splicing gene mutations in myeloid malignancies, with a high incidence of SRSF2 mutations in CMML.
Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. “Prognostic score including gene mutations in chronic myelomonocytic leukemia,”. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31(19):2428–36. The first and so far unique prognostic score to include molecular information.
Itzkson R, Fenaux P, Solary E. Chronic myelomonocytic leukemia: myelodysplastic or myeloproliferative? Best Pract Res Clin Haematol. 2013;26(4):387–400.
Aucagne R, Droin N, Paggetti J, Lagrange B, Largeot A, Hammann A, et al. “Transcription intermediary factor 1γ is a tumor suppressor in mouse and human chronic myelomonocytic leukemia,”. J Clin Invest. 2011;121(6):2361–70. The evidence that an epigenetically deregulated gene can drive a CMML phenotype.
Braun T, Itzykson R, Renneville A, de Renzis B, Dreyfus F, Laribi K, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood. 2011;118(14):3824–31.
Bou Samra E, Moreaux J, Vacheret F, Mills K, Rufflé F, Chiesa J, et al. New prognostic markers, determined using gene expression analyses, reveal two distinct subtypes of chronic myelomonocytic leukaemia patients. Br J Haematol. 2012;157(3):347–56.
Harrison CN, Mesa RA, Kiladjian J-J, Al-Ali H-K, Gisslinger H, Knoops L, et al. Health-related quality of life and symptoms in patients with myelofibrosis treated with ruxolitinib versus best available therapy. Br J Haematol. 2013;162(2):229–39.
Emanuel RM, Dueck AC, Geyer HL, Kiladjian J-J, Slot S, Zweegman S, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(33):4098–103.
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079–88.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–65.
Fenaux P, Beuscart R, Lai JL, Jouet JP, Bauters F. Prognostic factors in adult chronic myelomonocytic leukemia: an analysis of 107 cases. J Clin Oncol Off J Am Soc Clin Oncol. 1988;6(9):1417–24.
Morel P, Hebbar M, Lai JL, Duhamel A, Preudhomme C, Wattel E, et al. Cytogenetic analysis has strong independent prognostic value in de novo myelodysplastic syndromes and can be incorporated in a new scoring system: a report on 408 cases. Leukemia. 1993;7(9):1315–23.
Worsley A, Oscier DG, Stevens J, Darlow S, Figes A, Mufti GJ, et al. Prognostic features of chronic myelomonocytic leukaemia: a modified Bournemouth score gives the best prediction of survival. Br J Haematol. 1988;68(1):17–21.
Sanz GF, Sanz MA, Vallespí T, Cañizo MC, Torrabadella M, García S, et al. Two regression models and a scoring system for predicting survival and planning treatment in myelodysplastic syndromes: a multivariate analysis of prognostic factors in 370 patients. Blood. 1989;74(1):395–408.
Germing U, Strupp C, Aivado M, Gattermann N. “New prognostic parameters for chronic myelomonocytic leukemia,”. Blood. 2002;100(2):731–2. author reply 732–733.
Onida F, Kantarjian HM, Smith TL, Ball G, Keating MJ, Estey EH, et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99(3):840–9.
Germing U, Kündgen A, Gattermann N. Risk assessment in chronic myelomonocytic leukemia (CMML). Leuk Lymphoma. 2004;45(7):1311–8.
Patnaik MM, Padron E, LaBorde RR, Lasho TL, Finke CM, Hanson CA, et al. Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia. 2013;27(7):1504–10.
Such E, Germing U, Malcovati L, Cervera J, Kuendgen A, Della Porta MG, et al. “Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia,”. Blood. 2013;121(15):3005–15. The first scoring system dedicated to CMML that incorporates cytogenetics.
Grossmann V, Kohlmann A, Eder C, Haferlach C, Kern W, Cross NCP, et al. Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80 % of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia. 2011;25(5):877–9.
Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012;120(15):3080–8.
Damm F, Itzykson R, Kosmider O, Droin N, Renneville A, Chesnais V, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27(6):1401–3.
Kosmider O, Itzykson R, Chesnais V, Lasho T, Laborde R, Knudson R, et al. Mutation of the colony-stimulating factor-3 receptor gene is a rare event with poor prognosis in chronic myelomonocytic leukemia. Leukemia. 2013;27(9):1946–9.
M. M. Patnaik, R. Itzykson, T. L. Lasho, O. Kosmider, C. M. Finke, C. A. Hanson, R. A. Knudson, R. P. Ketterling, A. Tefferi, and E. Solary, “ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients,” Leukemia, Apr. 2014
Eissa H, Gooley TA, Sorror ML, Nguyen F, Scott BL, Doney K, et al. Allogeneic hematopoietic cell transplantation for chronic myelomonocytic leukemia: relapse-free survival is determined by karyotype and comorbidities. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant. 2011;17(6):908–15.
Park S, Labopin M, Yakoub-Agha I, Delaunay J, Dhedin N, Deconinck E, et al. Allogeneic stem cell transplantation for chronic myelomonocytic leukemia: a report from the Societe Francaise de Greffe de Moelle et de Therapie Cellulaire. Eur J Haematol. 2013;90(5):355–64.
Bacher U, Haferlach T, Schnittger S, Kreipe H, Kröger N. Recent advances in diagnosis, molecular pathology and therapy of chronic myelomonocytic leukaemia. Br J Haematol. 2011;153(2):149–67.
Cheng H, Kirtani VG, Gergis U. Current status of allogeneic HST for chronic myelomonocytic leukemia. Bone Marrow Transplant. 2012;47(4):535–41.
Koreth J, Pidala J, Perez WS, Deeg HJ, Garcia-Manero G, Malcovati L, et al. Role of reduced-intensity conditioning allogeneic hematopoietic stem-cell transplantation in older patients with de novo myelodysplastic syndromes: an international collaborative decision analysis. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31(21):2662–70.
J. Magenau and P. Reddy, “Next generation treatment of acute graft-versus-host disease,” Leukemia, Jun. 2014
Wattel E, Guerci A, Hecquet B, Economopoulos T, Copplestone A, Mahé B, et al. A randomized trial of hydroxyurea versus VP16 in adult chronic myelomonocytic leukemia. groupe Français des Myélodysplasies and European CMML group. Blood. 1996;88(7):2480–7.
Gerhartz HH, Marcus R, Delmer A, Zwierzina H, Suciu S, Dardenne M, et al. A randomized phase II study of low-dose cytosine arabinoside (LD-AraC) plus granulocyte-macrophage colony-stimulating factor (rhGM-CSF) in myelodysplastic syndromes (MDS) with a high risk of developing leukemia. EORTC Leukemia Cooperative Group. Leukemia. 1994;8(1):16–23.
Venditti A, Tamburini A, Buccisano F, Scimò MT, Del Poeta G, Maurillo L, et al. A phase-II trial of all trans retinoic acid and low-dose cytosine arabinoside for the treatment of high-risk myelodysplastic syndromes. Ann Hematol. 2000;79(3):138–42.
Nair R, Nair CN, Advani SH. All trans retinoic acid with low dose cytosine arabinoside in the treatment of myelodysplastic syndrome. Leuk Lymphoma. 1998;29(1–2):187–92.
Lancet EJ, Gojo I, Gotlib J, Feldman EJ, Greer J, Liesveld JL, et al. A phase 2 study of the farnesyltransferase inhibitor tipifarnib in poor-risk and elderly patients with previously untreated acute myelogenous leukemia. Blood. 2007;15(109):1387–94.
Beran M, Estey E, O’Brien S, Cortes J, Koller CA, Giles FJ, et al. Topotecan and cytarabine is an active combination regimen in myelodysplastic syndromes and chronic myelomonocytic leukemia. J Clin Oncol Off J Am Soc Clin Oncol. 1999;17(9):2819–30.
Griffiths EA, Gore SD. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin Hematol. 2008;45(1):23–30.
Wijermans PW, Rüter B, Baer MR, Slack JL, Saba HI, Lübbert M. Efficacy of decitabine in the treatment of patients with chronic myelomonocytic leukemia (CMML). Leuk Res. 2008;32(4):587–91.
Scott BL, Ramakrishnan A, Storer B, Becker PS, Petersdorf S, Estey EH, et al. Prolonged responses in patients with MDS and CMML treated with azacitidine and etanercept. Br J Haematol. 2010;148(6):944–7.
Adès L, Sekeres MA, Wolfromm A, Teichman ML, Tiu RV, Itzykson R, et al. Predictive factors of response and survival among chronic myelomonocytic leukemia patients treated with azacitidine. Leuk Res. 2013;37(6):609–13.
T. Braun (Groupe français des myelodysplasies, Bobigny, France). “Mutational analysis and long term outcome in advanced chronic myelomonocytic leukemia (CMML) treated by decitabine : an update of the GFM-CMML-2007 phase II trial.,” 19th EHA congress, 2014
T. Svensson, O. Chowdhury, H. Garelius, F. Lorenz, L. Saft, S.-E. Jacobsen, E. Hellström-Lindberg, and H. Cherif, “A pilot phase I dose finding safety study of the thrombopoietin-receptor agonist, eltrombopag, in patients with myelodysplastic syndrome treated with azacitidine,” Eur. J. Haematol., May 2014
Basciano PA, Bussel JB. Thrombopoietin-receptor agonists. Curr Opin Hematol. 2012;19(5):392–8.
Giagounidis A, Mufti GJ, Fenaux P, Sekeres MA, Szer J, Platzbecker U, et al. Results of a randomized, double-blind study of romiplostim versus placebo in patients with low/intermediate-1-risk myelodysplastic syndrome and thrombocytopenia. Cancer. 2014;120(12):1838–46.
Bachegowda L, Gligich O, Mantzaris I, Schinke C, Wyville D, Carrillo T, et al. Signal transduction inhibitors in treatment of myelodysplastic syndromes. J Hematol Oncol J Hematol Oncol. 2013;6:50.
Oshima Y, Yuji K, Tanimoto T, Hinomura Y, Tojo A. Association between acute myelogenous leukemia and thrombopoietin receptor agonists in patients with immune thrombocytopenia. Intern Med Tokyo Jpn. 2013;52(19):2193–201.
Compliance with Ethics Guidelines
Conflict of Interest
Dr. Dorothée Selimoglu-Buet and Dr. Eric Solary each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Selimoglu-Buet, D., Solary, E. Chronic Myelomonocytic Leukemia Prognostic Classification and Management: Evidence Base and Current Practice. Curr Hematol Malig Rep 9, 301–310 (2014). https://doi.org/10.1007/s11899-014-0225-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-014-0225-2