
Abstract
Purpose of Review
Diabetic retinopathy (DR), a leading cause of visual impairment in the developed country, is characterized by vascular lesions and neuronal damage of the retina. Treatment options for this condition are currently limited. The advent of therapy targeting vascular endothelial growth factor (VEGF) demonstrated significant benefits to patients with DR. However, this treatment is limited by its short half-life and requirement for frequent invasive intravitreal injections. In addition, many patients failed to achieve clinically significant improvement in visual function. Gene therapy has the potential to provide an alternative treatment for DR with distinct advantages, such as longer therapeutic effect, less injection frequency, ability to intervene at disease onset, and potentially fewer side effects.
Recent Findings
Strategies for gene therapy in DR, stemming from the current understanding of the disease pathogenesis, focus on the inhibition of neovascularization and protection of neurovascular degeneration in the retina. Studies with promising results have mainly focussed on animal models due to efficacy and safety concerns, despite a number of successful preclinical studies using adeno-associated virus-mediated transduction to treat both vascular dysfunction and neuronal degeneration. With the optimization of delivery vectors, transgene regulation, and outcome measure, gene therapy will potentially become available for patients with DR.
Summary
This review provides an update on the current strategies utilized in DR gene therapy research. Several barriers to the clinical application of gene therapy for DR are highlighted, and future directions for this research are proposed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gene therapy is designed to introduce genetic material into the cells of patients to compensate faulty genes or deliver therapeutic transgenes. Over the past three decades, gene therapy has progressed from initial human gene transfer experiments to approved clinical therapies [1]. Successful gene therapy trials have led to approval of a number of gene therapy products by drug regulation bodies across Europe and the USA. These include Glybera (alipogene tiparvovec), a recombinant adeno-associated virus (rAAV) product for the treatment of lipoprotein lipase deficiency in 2012 [2], and Exondys51 (eteplirsen), an antisense oligonucleotide compound to restore the reading frame in patients with Duchenne muscular dystrophy in 2016 [3]. In 2017, the US Food and Drug Administration (FDA) approved gene therapy product, Luxturna (voretigene neparvovec), which is an adeno-associated virus (AAV)-based gene therapy for the inherited retinal disease RPE65-associated Leber’s congenital amaurosis (LCA) [4], marking an historic landmark in gene therapy for eye diseases. Since the efficacy and safety of AAV-based gene therapy for hereditary eye diseases has been established in clinical trials, interest has grown in the use of gene therapy to treat complex polygenic eye disorders such as glaucoma, age-related macular degeneration (AMD), and diabetic retinopathy (DR). DR is an increasingly prevalent chronic condition with complex pathogenesis that is incompletely understood. Gene therapy interventions could potentially provide longer-term treatment with fewer side effects than current therapeutics; however, there are still several barriers to overcome before such a DR treatment could reach clinical trials. This review will provide an overview of the development of gene therapies for DR.
Pathophysiology and Treatments of Diabetic Retinopathy
DR is a common complication of diabetes mellitus and one of the leading causes of blindness among working-age people in the developed country. Severe stages of DR include proliferative diabetic retinopathy (PDR), resulting from abnormal growth of new retinal blood vessels, and diabetic macular edema (DME), where retinal vascular leakage and edema occur in the central part of the retina [5]. It is estimated that there are 17 million and 21 million patients with PDR and DME, respectively [6]. With an increased prevalence of diabetes from 415 million in 2015 to nearly 642 million by 2040, the prevalence of DR is expected to rise accordingly [7].
Control of blood glucose in patients with diabetes is essential to prevent or ameliorate progression of DR; however, beyond this, treatment for severe DR is limited to a few modalities, such as photocoagulation for PDR and anti-vascular endothelial growth factor (VEGF) for DME. Although these treatments are available, no treatment is yet to fully attenuate clinical progression to reverse damage to the retina. Our previous review detailed several canonical biochemical pathways triggered by hyperglycemia in DR [8]. Despite microcirculatory impairment traditionally being regarded as the main feature of DR, emerging evidence clearly shows retinal neurodegeneration occurring before clinical signs of DR, with subsequent microvascular dysfunction [9,10,11]. A growing body of evidence highlights the significant role of retinal neuronal dysfunction and death in DR, causing functional deficits such as decreased dark adaptation, contrast sensitivity, color perception, and visual field [10, 12, 13].
Current Progress in Gene Therapy for Diabetic Retinopathy
The goal of gene therapy is to achieve adequate expression of a transgene at a level to decrease or cure disease condition with minimal adverse effects. There are several strategies of gene therapy, including gene augmentation, gene-specific targeting, and most recently, genome editing [14]. Gene augmentation therapy introduces a new functional gene into the host cell to compensate for a faulty gene, which has mostly been applied to monogenic disease. Gene-specific targeted therapy is designed to alter the activity of an existing abnormal gene, or to provide an intact copy of a gene for protection or regeneration. Genome editing or correction therapy directly repairs a mutant gene into a functional gene which thereby can fundamentally correct mutant genes.
Current investigations into gene therapy for DR utilize gene-specific targeted therapy divided into two approaches according to disease pathophysiology: those that target existing neovascularization and vascular hyperpermeability, and those that aim to protect retinal blood vessels and neurons from damage. Within these categories, a variety of candidate genes have been studied as potential targets for DR gene therapy.
Targeting Retinal Vasculopathy of Diabetes
Increased vascular permeability caused by hyperglycemia results in accumulation of intraretinal fluid and other manifestations, such as intraretinal hard exudates and cotton-wool spots, which are the main features of DME. PDR is characterized by retinal neovascularization in late-stage DR, due to endothelial cell proliferation and imbalance between pro-angiogenic and antiangiogenic factors. Accordingly, one of the approaches to DR gene therapy is to inhibit proliferation of endothelial cells or promote anti-angiogenesis (Table 1).
Treatment for both DME and PDR has been revolutionized by the availability of intravitreal injection of anti-VEGF agents. Such agents bind VEGF therefore preventing VEGF pathway and suppressing neovascularization [29]. However, there are shortcomings with regard to variable response to therapy and lack of efficacy in a subgroup of patients. VEGF is an evident therapeutic target for DR gene therapy studies. Various experiments have attempted to interfere in the intraocular VEGF pathway, both extra- and intracellularly. sFlt-1, a soluble splice variant of the VEGF receptor 1 (VEGFR-1 or Flt-1), acts as a decoy VEGF receptor in the extracellular space, and has been investigated as a potential target to suppress retinal neovascularization in experimental animal models [19, 22, 23, 26, 28, 30]. Flt23k, an anti-VEGF intraceptor, is a recombinant construct of VEGF binding domains 2 and 3 of VEGFR-1 coupled with the endoplasmic reticulum (ER) retention signal sequence lysine-aspartic acid-glutamic acid-leucine (KDEL) [29]. Flt23k can thus intervene VEGF pathways and degrade VEGF intracellularly. A study reported a clear reduction of choroidal neovascularization in an animal model after injection of AAV-mediated Flt23k, providing indirect evidence of potential of AAV-Flt23k in impeding retinal neovascularization [31]. Future studies targeting VEGF with AAV as a delivery vector should examine its efficacy and safety in animal models with DR. Other strategies, such as the injection of siRNA-expressing plasmids targeting VEGF, have also showed an effective reduction of VEGF expression, inhibiting retinal neovascularization in animals in early days of gene therapy [21].
Another approach to suppress retinal angiogenesis is to introduce endogenous angiogenesis inhibitors, such as pigment epithelium-derived factor (PEDF), angiogestin, endostatin, tissue inhibitor metalloproteinase-3, and calreticulin antiangiogenic domain (CAD). A study using AAV to deliver PEDF in the retina demonstrated decreased VEGF levels and reduced expression of angiogenesis-related fibrosis factors, such as matrix metalloproteinases and connective tissue growth factor in transgenic mice designed to develop progressive neovascularization [18]. Angiostatin is a fragment of plasminogen with the capability of inhibiting endothelial proliferation [32]. In a murine model of oxygen-induced retinopathy (OIR), a rodent model of retinal neovascularization, lentiviral vector-encoded angiostatin was shown to be a potent inhibitor of neovascularization, although therapeutic safety was not examined [25]. Like angiostatin, endostatin is proteolytic fragment of collagen XVIII and a potential therapeutic target for neovascularization owing to its antiangiogenic properties [33]. An earlier study injecting endostatin molecules in animals exposed to OIR showed a marked decrease of VEGF and integrin β3 levels, both of which are strongly related to angiogenesis [34]. In addition, retinal vascular architecture has been partly preserved in those mice injected with AAV expressing endostatin [17]. Retinal transduction of AAV encoding other endogenous angiogenesis inhibitors such as tissue inhibitor metalloproteinase-3 or CAD has also been found to reduce retinal neovascularization in OIR mice [16, 27].
Molecules enabling inhibition of endothelial cell proliferation, such as amino-terminal fragment (ATF) [35], and kringle 1 domain of hepatocyte growth factor (HGFK1) [36], have been increasingly investigated as potential therapeutic target of gene therapy for DR. Amino-terminal fragment (ATF) derived from urokinase-type plasminogen activator impedes the migration of endothelial cells inhibiting urokinase-type plasminogen activator binding to its receptor [37]. Adenovirus-mediated delivery of ATF or endostatin showed a reduction of retinal neovascularization in OIR mice, but the safety of using adenovirus was not investigated [24]. HGFK1 can suppress proliferation of tumor cells in vitro and vascular cell growth in an animal tumor model [38], indicating HGFK1 may be able to inhibit ocular angiogenesis. A recent study reported that retinal transduction of AAV-mediated HGFK1 could maintain normal vascular architecture and reduced VEGF expression in the retina of an OIR mouse model [15].
Inhibitors of vascular permeability, such as sFlt-1 and vasoinhibin, are desirable therapeutic transgene candidates for DR gene therapy. Retinal transduction of AAV encoding sFlt-1 and vasoinhibin can reduce blood-retinal barrier breakdown and decrease microvascular abnormalities, as well as restore some retinal functions after diabetes was induced by streptozotocin (STZ) in rats [39, 40]. Interestingly, AAV transduction efficacy was found to be increased in STZ-induced diabetic rats compared to controls, which is likely due to elevated cell entry [39]. Another study also reported that AAV2-vasoinhibin vectors are relatively more efficient than AAV2-sFlt-1 vectors in terms of reduction of vasopermeability in diabetic rats [40].
Vascular and Neuronal Protection
In addition to suppression of existing abnormal blood vessels in retina, gene therapy for DR could be applied as an intervention for the vascular and neuronal degeneration in the early stages of DR, prior to obvious clinical pathologies. An improved understanding of pathophysiology of DR has precipitated studies into the prevention of retinal vascular dysfunction, neuronal apoptosis, or reducing oxidative stress (Table 2).
Early growth response 1 (EGR1) is an important zinc finger transcription factor involved in diverse pathways. Hyperglycemia in diabetes causes a dramatic increase of EGR1 expression in vascular cells, which may be one of the key initial events in the early development of DR. Inhibition of EGR1 expression may reduce injurious effects of hyperglycemia on retinal vascular function. An in vivo study using an AAV vector encoding small hairpin RNAs (artificial RNA molecules that can be used to silence target gene expression via RNA interference) directed against EGR1 showed retinal transduction of these vectors dramatically reduced apoptosis in cells localized to the inner nuclear layer and outer nuclear layer [41]. The study confirmed that EGR1 regulated retinal endothelial cell function, such as migration and vascularization, via the induction of p53 expression [41].
Autophagy, a conserved metabolic process, is characterized by lysosomal degradation of dysfunctional proteins or organelles. Studies have shown that microtubule-associated protein 1 light chain 3-II (LC3B-II) is associated with the extent of autophagosome formation and act as a significant molecular biomarker to detect autophagic activity [49, 50]. In diabetic rats, LC3B-II was suppressed by high glucose levels, suggesting an increase of LC3B might impair DR progression. Intravitreal injection of diabetic rats with adenovirus vectors encoding anti-microRNA-204 enhanced the expression of LC3B-II and promoted autophagy, minimizing the detrimental development of DR [42].
Vascular and neural cells in DR are subject to apoptosis, at least partly due to upregulation of the membrane attack complex [51]. Soluble CD59 (sCD59), a membrane-independent inhibitor of membrane attack complex, protects the blood-retinal barrier and retinal neurons from damage. In an STZ-induced diabetic mouse model, sCD59 delivered by AAV reduced vascular leakage by 60% compared with control mice [46]. Furthermore, sCD59 has been demonstrated to activate retinal glial cells in the short-term. However, concerns remain about whether long-term expression of sCD59 would result in reactive gliosis, a pathogenic process causing neurodegeneration [46].
The application of neurotrophic factors is another neuroprotective approach for DR gene therapy. Intravitreal injection of AAV vectors encoding brain-derived neurotrophic factor (BDNF) demonstrated an increased survival rate of retinal ganglion cells and improved retinal function in STZ-induced diabetic rats compared to controls [48]. Overexpression of erythropoietin (EPO), a hematopoietic cytokine, has also been shown to have a potent neuroprotective effect in DR. One attempt demonstrated that intravitreal injection of EPO protects against retinal vascular regression at the early stage of DR in STZ-induced diabetic rats through its downregulations on EPO receptor, VEGF, and VEGF receptor [52]. Subretinal AAV2-mediated expression of EPO was shown to be successful in preventing blood-retinal barrier disruption and retinal neuronal apoptosis in STZ-induced diabetic rats [45]. These studies suggest that the use of neurotrophic factors could elicit positive effects in the early stages of DR and warrants further investigation on the need for preventing neurodegeneration.
Oxidative stress is an important event in the pathogenesis in DR, making it an obvious target for gene therapy studies [53]. In a diabetic rat model, retinal transduction of AAV-mediated manganese superoxide dismutase (MnSOD), the key antioxidant enzyme in mitochondria, has been shown to reduce basement membrane thickening and retinal capillary apoptosis by enhancing the endogenous level and activity of retinal MnSOD and its catalase [44]. Moreover, these AAV vectors also effectively prevented the progression of DR and the metabolic memory phenomenon, wherein hyperglycemia following the onset of diabetes is “remembered” despite subsequent attainment of improved glycemic control [44].
A growing body of evidence implicates the involvement of the local renin-angiotensin system (RAS) in retinal vascular permeability and oxidative stress. Clinical studies have clearly shown the benefits of RAS inhibition in both type 1 and type 2 diabetic patients with retinopathy [54]. Nevertheless, current RAS inhibitors could not completely reverse neurodegeneration and vascular dysfunction due to the presence of local angiotensin II (Ang II). Moreover, the effect of suppression of RAS via angiotensin-converting enzymes and Ang II type 1 antagonists in the retina have yet to be confirmed. One study investigated a counter-regulatory and vasoprotective axis involving angiotensin-converting enzyme 2 (ACE2), angiotensin-(1–7) (Ang-(1–7)), and the Mas receptor [55]. Injection of AAV vectors encoding ACE2 and Ang-(1–7) resulted in significant reduction of diabetes-induced retinal vascular leakage, acellular capillaries, inflammation, and oxidative stress in diabetes-induced rodents [43, 47]. These findings suggest that enhancing the ACE2/Ang-(1–7) axis of RAS could protect diabetic retina from progressive vascular dysfunction and neurodegeneration.
CRISPR/Cas-Based Therapies
The invention of CRISPR/Cas system has allowed gene therapy to progress from gene augment to gene editing. Using single-guide RNAs, CRISPR/Cas recognizes a specific DNA sequence that is subsequently cut by a nuclease such as Cas9. This technology has the potential to be applied in gene therapy for DR targeting the over-expressed VEGF. CRISPR/Cas systems carrying different nucleases, such as Cas9 and Cpf1, along with small guide RNAs that target VEGF and hypoxia inducing factor 1a (Hif1a), have been used to effectively prevent choroidal neovascularization in a mouse model of AMD [56, 57]. Other forms of modified CRISPR/Cas systems where nuclease loses its cutting ability but binds with specific sequence under the guidance of sgRNA were developed to activate and inhibit gene expression in CRISPRi (CRISPR interference) and CRISPRa (CRISPR activation), respectively [58]. These new CRISPR/Cas systems could be exploited in potential DR treatments that activate gene expression of antiangiogenic factors like PEDF, or alternatively suppress expression of angiogenic factors such as VEGF.
Overall, studies utilizing gene therapy targeting pathological changes in the retina in early and late stage DR have yet to lead to a commercially available treatment. CRISPR/Cas has been an effective tool in animal trials for AMD and may prove similarly suited to DR therapies. Despite encouraging some experimental results targeting either retinal vasculopathy or neurovascular protection, many limitations and challenges are currently hindering the translation of gene therapy for DR from the laboratory to clinical applications.
Clinical Trial Outcomes for Other Ocular Diseases
Gene therapy clinical trials (Table 3) for DME were conducted a decade ago using bevasiranib, a small interfering RNA (siRNA) pharmaceutical targeting VEGF [59]. Although bevasiranib was initially reported as safe and effective, a phase 3 trial with combination of bevasiranib and ranibizumab (a commercially available anti-VEGF reagent) for wet AMD patients was terminated after 2 years on the recommendation of the study’s Independent Data Monitoring Committee due to safety and efficacy concerns [60]. A critical in vitro study conducted 4 years earlier showed evidence of siRNA-induced off-target effects, generating a measurable phenotype [61]. Published clinical trial data for the treatment of AMD using bevasiranib produced consistent short-term outcomes, but the lack of long-term endpoints means the lasting efficacy of this treatment is not yet established. Compared with none of clinical trials for gene therapy for DR, there are several closed and ongoing trials designed for wet AMD where AAV is used as the delivery vector. Previous gene therapy trials for wet AMD were discontinued due to inconsistent efficacy, but importantly, they reported no safety concerns [62]. The understanding of gene therapy for inherited retinal disease amenable to gene therapy has advanced significantly with time. Improvements in viral vectors, better selection of transgenes, refined surgical skills to deliver vectors and advanced outcome measures may improve outcomes in future clinical trials for non-inherited eye diseases.
Challenges in Gene Therapy for Diabetic Retinopathy
While the landmark gene therapy product, Luxturna, for treatment of Leber’s congenital amaurosis, was approved by FDA in 2017 [63], gene therapies for generic eye disease, such as DR, are still in their infancy due to several factors that hinder clinical translation. Major contributing factors include the complex pathogenesis of the disease itself, issues related to the safety and specificity of the vector delivery of transgenes, patient selection for clinical trials and standardization of outcome measurements.
Limited Understanding of DR Pathogenesis
Limited understanding of DR pathogenesis poses a major obstacle for applying gene therapy at a clinical level. In contrast to many gene therapies in clinical trials that target monogenic eye disease, DR is a polygenetic and relatively heterogeneous disorder. This means gene therapy for DR may need to target a variety of gene candidates that are implicated in the pathogenesis of DR. Additionally, DR is a progressive disease with several staging classifications based on the severity of the symptoms over the disease course, which convolutes patient selection criteria. Furthermore, it may not be appropriate for participants with the same clinical classification to undertake the same therapy given the diverse range of genetic polymorphisms that can contribute to a similar clinical picture. This complexity starkly differs from the relative simplicity of the pathogenesis of LCA, an inherited retinal dystrophy attributable to mutations in a single gene, RPE65, with clinical features evident from birth and for which there is currently gene therapy commercially available [63]. The complicated pathogenesis of DR contributes to the difficulty of creating an appropriate animal model, which further hinders the translation of gene therapy research for this condition.
With the application of new techniques such as single cell sequencing and CRISPR/Cas gene editing technology, our understanding of the pathogenesis of DR is evolving as new genes associated with different stages of DR are discovered. While VEGF has been a key therapeutic target, these new techniques will allow other genes involved in critical pathways in DR to be identified and validated for gene therapy.
Vector Safety and Stability
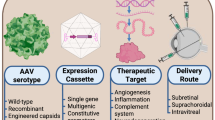
Viral vectors pose another major concern for an effective and safe delivery of transgenes in gene therapy. Over the past two decades, studies have increasingly used AAV instead of adenovirus or lentivirus in both basic and clinical research of gene therapy for eye diseases. Compared with other viral vectors, AAV is less immunogenic and only a small portion undergoes genome integration, although major limitations include its relatively small transgene packaging capacity and high cost for clinical-grade manufacturing (Table 4). Our previous review has detailed the differences between these vector modalities [8]. AAV2 is currently the most-used viral vector in gene therapy clinical trials; however, its transduction efficacy in the retina is limited, and its target cell-type depends on the administration route due to physical and cellular barriers; for example, photoreceptors are transduced via subretinal injection whilst ganglion cells are transduced via intravitreal injection [64]. Müller cells, a key source of VEGF expression in DME and PDR, cross through all layers of retina, which are not well transduced by AAV2 with ubiquitous promoters, such as cytomegalovirus, chicken β-actin or cytomegalovirus early enhancer/chicken β-actin [8]. Glial fibrillary acidic protein promoter, a glial cell-specific promoter that modulates transgene expression in AAV2, has been demonstrated to target Müller cells specifically and to supress retinal neovascularization significantly in OIR models [17].
Alternatively, AAV vectors can be modified to improve their viability. An AAV variant, 7m8, developed by in vivo directed evolution, demonstrated stronger transduction in the outer retina compared with AAV2 [65]. Using in silico analysis, Anc80, a predicted AAV ancestor, showed a robust increase in transduction efficacy than AAV2 [66]. In contrast to conventional AAV, self-complementary AAV (scAAV) encompasses an inverted repeat gene sequence that is able to fold into a section of double-stranded DNA without the synthesis of a second DNA strand. An in vitro study showed the transduction efficacy of scAAV is 5- to 140-fold higher than conventional AAV [67]. However, scAAV can only hold about half of that packaging capacity of conventional AAV vector (~ 2.4 kb for scAAV vs ~ 4.7 kb for conventional AAV) [67].
The deficiencies of AAV as a viral vector have led to the development of non-viral vectors. Synthetic vectors tend to show mild immune responses in part due to the lack of preexisting antibodies and can carry large transgene payloads. Their production for clinical application is easier and cheaper than traditional viral vectors [68]. Despite these advantages, non-viral vectors have been largely confined to experimental settings because of their poor transduction efficacy comparing with viral vectors [8].
Targeted Modulation of Transgene Expression
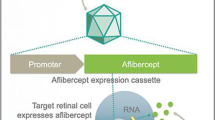
Another challenge for gene therapy is to ensure that only those targeted cells or tissues are effectively transduced, as unintentional transduction of non-targeted cells could potentially cause toxicity or adverse reactions. All vectors contain an expression cassette where transgene and regulatory sequence, such as promoters, are included. Promoters can regulate transgene expression and determine target specificity. Most studies use ubiquitous promoters in vector design, such as cytomegalovirus, chicken β-actin and its derivative cytomegalovirus early enhancer/chicken β-actin, so that most cells are transduced without target specificity. Future identification of promoters that target specific cell types would minimize off-target effects of viral vectors, and lead to safer clinical safety profiles.
Ideally, transgene expression could be modulated according to disease activity. A few of studies have shown that gene expression can be switched on and off at the protein level through the utility of a destabilizing domain [69, 70]. The destabilizing domain-fused protein of interest can be degraded by the endogenous proteasomal systems, and when the disease is in its active stage, the fused protein can be rescued in the presence of shielding small molecule ligands such as synthetic ligand shield-1, trimethoprim, and 4-hydroxytamoxifen [71]. Other approaches to regulate transgene expression may include specific enhancer or responding elements incorporated in the expression cassette, for instance enhancers bound to transcription factors upregulated during active DR progression, such as HIF-1 [72]. MicroRNA responding elements could also be used to regulate transgene expression at stable and safe levels [73].
Patient Selection and Protocol for Outcome Measurement in Clinical Trials
Patient selection for a clinical trial of gene therapy for DR would need to account for a number of factors. Only individuals with a history of strict glycemic control who are responsive to conventional treatment should be selected as potential candidates due to the metabolic memory phenomenon. Detectable genetic and epigenetic risk factors also need to be controlled for in any future trials.
To assess and monitor the efficacy and safety of gene therapy in clinical trials, the measurement of treatment outcomes is a key consideration. Reproducible and reliable testing of outcome measurements are required to justify clinical trial data for ocular gene therapies [74]. Visual acuity (VA) is a widely accepted measure methodology in 94% of gene therapy trials. However, higher variability in VA has been identified in low vision patients and different VA measures were reported even within the same trials for the same disease, indicating that optimization of VA measurements should be studied further [75]. A review of ocular gene therapy clinical trial publications suggested that formal natural history studies and additional validation studies of novel outcome measures should be included in an effort to standardize outcomes. The outcome measures recorded need to be optimized according to the specific molecular target location within the eye. Moreover, appropriate outcome measures being specific to DR, such as Early Treatment Diabetic Retinopathy Study (ETDRS) scale [76] and diabetic retinopathy severity score [77], should also be taken into account for a thorough review for treatment efficacy and safety.
Conclusion
The FDA landmark approval of Luxturna for LCA was a significant milestone for gene therapy. Whilst most of the Phase III gene therapy clinical trials currently in progress target inherited eye diseases, an increasing number of studies are now investigating complex ocular diseases including AMD and DR. Gene therapy approaches to DR can be broadly categorized as either targeting the pro- and antiangiogenic factors involved in neovascularization in late-stage DR, or preventing vascular and neuronal degeneration in the early stages of DR. The barriers to clinical trials are due to the complexity of DR pathogenesis, patient selection and outcome measurement, as well as broader challenges pertaining to vector safety, selection, targeting, and modulation.
References
Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science. 1995;270(5235):475–80. https://doi.org/10.1126/science.270.5235.475.
Yla-Herttuala S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol Ther. 2012;20(10):1831–2. https://doi.org/10.1038/mt.2012.194.
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351(6268):84–8. https://doi.org/10.1126/science.aad5227.
Smalley E. First AAV gene therapy poised for landmark approval. Nat Biotechnol. 2017;35(11):998–9. https://doi.org/10.1038/nbt1117-998.
Wong TY, Cheung CM, Larsen M, Sharma S, Simo R. Diabetic retinopathy. Nat Rev Dis Primers. 2016;2:16012. https://doi.org/10.1038/nrdp.2016.12.
Yau JW, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35(3):556–64. https://doi.org/10.2337/dc11-1909.
Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, Cho NH, et al. IDF diabetes atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract. 2017;128:40–50. https://doi.org/10.1016/j.diabres.2017.03.024.
Wang JH, Ling D, Tu L, van Wijngaarden P, Dusting GJ, Liu GS. Gene therapy for diabetic retinopathy: are we ready to make the leap from bench to bedside? Pharmacol Ther. 2017;173:1–18. https://doi.org/10.1016/j.pharmthera.2017.01.003.
Abcouwer SF, Gardner TW. Diabetic retinopathy: loss of neuroretinal adaptation to the diabetic metabolic environment. Ann N Y Acad Sci. 2014;1311:174–90. https://doi.org/10.1111/nyas.12412.
Simo R, Hernandez C. European Consortium for the Early Treatment of Diabetic R. Neurodegeneration in the diabetic eye: new insights and therapeutic perspectives. Trends Endocrinol Metab. 2014;25(1):23–33. https://doi.org/10.1016/j.tem.2013.09.005.
Simo R, Hernandez C. Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog Retin Eye Res. 2015;48:160–80. https://doi.org/10.1016/j.preteyeres.2015.04.003.
Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52(2):1156–63. https://doi.org/10.1167/iovs.10-6293.
Du Y, Veenstra A, Palczewski K, Kern TS. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc Natl Acad Sci U S A. 2013;110(41):16586–91. https://doi.org/10.1073/pnas.1314575110.
Lee JH, Wang JH, Chen J, Li F, Edwards TL, Hewitt AW, et al. Gene therapy for visual loss: opportunities and concerns. Prog Retin Eye Res. 2019;68:31–53. https://doi.org/10.1016/j.preteyeres.2018.08.003.
Sun P, Liu Z. Overexpressing kringle 1 domain of hepatocyte growth factor with adeno-associated virus inhibits the pathological retinal neovascularization in an oxygen-induced retinopathy mouse model. Biochem Biophys Res Commun. 2019;508(1):130–7. https://doi.org/10.1016/j.bbrc.2018.11.111.
Tu L, Wang JH, Barathi VA, Prea SM, He Z, Lee JH, et al. AAV-mediated gene delivery of the calreticulin anti-angiogenic domain inhibits ocular neovascularization. Angiogenesis. 2018;21(1):95–109. https://doi.org/10.1007/s10456-017-9591-4.
Biswal MR, Prentice HM, Dorey CK, Blanks JC. A hypoxia-responsive glial cell-specific gene therapy vector for targeting retinal neovascularization. Invest Ophthalmol Vis Sci. 2014;55(12):8044–53. https://doi.org/10.1167/iovs.14-13932.
Haurigot V, Villacampa P, Ribera A, Bosch A, Ramos D, Ruberte J, et al. Long-term retinal PEDF overexpression prevents neovascularization in a murine adult model of retinopathy. PLoS One. 2012;7(7):e41511. https://doi.org/10.1371/journal.pone.0041511.
Pechan P, Rubin H, Lukason M, Ardinger J, DuFresne E, Hauswirth WW, et al. Novel anti-VEGF chimeric molecules delivered by AAV vectors for inhibition of retinal neovascularization. Gene Ther. 2009;16(1):10–6. https://doi.org/10.1038/gt.2008.115.
Lai CM, Shen WY, Brankov M, Lai YK, Barnett NL, Lee SY, et al. Long-term evaluation of AAV-mediated sFlt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol Ther. 2005;12(4):659–68.
Jiang J, Xia XB, Xu HZ, Xiong Y, Song WT, Xiong SQ, et al. Inhibition of retinal neovascularization by gene transfer of small interfering RNA targeting HIF-1alpha and VEGF. J Cell Physiol. 2009;218(1):66–74. https://doi.org/10.1002/jcp.21566.
Lamartina S, Cimino M, Roscilli G, Dammassa E, Lazzaro D, Rota R, et al. Helper-dependent adenovirus for the gene therapy of proliferative retinopathies: stable gene transfer, regulated gene expression and therapeutic efficacy. J Gene Med. 2007;9(10):862–74. https://doi.org/10.1002/jgm.1083.
Ideno J, Mizukami H, Kakehashi A, Saito Y, Okada T, Urabe M, et al. Prevention of diabetic retinopathy by intraocular soluble flt-1 gene transfer in a spontaneously diabetic rat model. Int J Mol Med. 2007;19(1):75–9.
Le Gat L, Gogat K, Bouquet C, Saint-Geniez M, Darland D, Van Den Berghe L, et al. In vivo adenovirus-mediated delivery of a uPA/uPAR antagonist reduces retinal neovascularization in a mouse model of retinopathy. Gene Ther. 2003;10(25):2098–103. https://doi.org/10.1038/sj.gt.3302122.
Igarashi T, Miyake K, Kato K, Watanabe A, Ishizaki M, Ohara K, et al. Lentivirus-mediated expression of angiostatin efficiently inhibits neovascularization in a murine proliferative retinopathy model. Gene Ther. 2003;10(3):219–26. https://doi.org/10.1038/sj.gt.3301878.
Gehlbach P, Demetriades AM, Yamamoto S, Deering T, Xiao WH, Duh EJ, et al. Periocular gene transfer of sFlt-1 suppresses ocular neovascularization and vascular endothelial growth factor-induced breakdown of the blood-retinal barrier. Hum Gene Ther. 2003;14(2):129–41. https://doi.org/10.1089/104303403321070829.
Auricchio A, Behling KC, Maguire AM, O'Connor EM, Bennett J, Wilson JM, et al. Inhibition of retinal neovascularization by intraocular viral-mediated delivery of anti-angiogenic agents. Mol Ther. 2002;6(4):490–4. https://doi.org/10.1006/mthe.2002.0702.
Bainbridge JW, Mistry A, De Alwis M, Paleolog E, Baker A, Thrasher AJ, et al. Inhibition of retinal neovascularisation by gene transfer of soluble VEGF receptor sFlt-1. Gene Ther. 2002;9(5):320–6. https://doi.org/10.1038/sj.gt.3301680.
Prea SM, Chan EC, Dusting GJ, Vingrys AJ, Bui BV, Liu GS. Gene therapy with endogenous inhibitors of angiogenesis for neovascular age-related macular degeneration: beyond anti-VEGF therapy. J Ophthalmol. 2015;2015:201726–12. https://doi.org/10.1155/2015/201726.
Lai YK, Shen WY, Brankov M, Lai CM, Constable IJ, Rakoczy PE. Potential long-term inhibition of ocular neovascularisation by recombinant adeno-associated virus-mediated secretion gene therapy. Gene Ther. 2002;9(12):804–13. https://doi.org/10.1038/sj.gt.3301695.
Zhang X, Das SK, Passi SF, Uehara H, Bohner A, Chen M, et al. AAV2 delivery of Flt23k intraceptors inhibits murine choroidal neovascularization. Mol Ther. 2015;23(2):226–34. https://doi.org/10.1038/mt.2014.199.
O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79(2):315–28. https://doi.org/10.1016/0092-8674(94)90200-3.
O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277–85. https://doi.org/10.1016/s0092-8674(00)81848-6.
Zhang M, Yang Y, Yan M, Zhang J. Downregulation of vascular endothelial growth factor and integrinbeta3 by endostatin in a mouse model of retinal neovascularization. Exp Eye Res. 2006;82(1):74–80. https://doi.org/10.1016/j.exer.2005.05.005.
Kim KS, Hong YK, Lee Y, Shin JY, Chang SI, Chung SI, et al. Differential inhibition of endothelial cell proliferation and migration by urokinase subdomains: amino-terminal fragment and kringle domain. Exp Mol Med. 2003;35(6):578–85. https://doi.org/10.1038/emm.2003.76.
Xin L, Xu R, Zhang Q, Li TP, Gan RB. Kringle 1 of human hepatocyte growth factor inhibits bovine aortic endothelial cell proliferation stimulated by basic fibroblast growth factor and causes cell apoptosis. Biochem Biophys Res Commun. 2000;277(1):186–90. https://doi.org/10.1006/bbrc.2000.3658.
Sun Z, Zhang PX, Wang P, Gurewich V, Shen HY, Liu JN. Amino-terminal fragment of urokinase-type plasminogen activator inhibits its plasminogen activation. Thromb Res. 2002;106(2):105–11. https://doi.org/10.1016/s0049-3848(02)00091-9.
Shen Z, Yang ZF, Gao Y, Li JC, Chen HX, Liu CC, et al. The kringle 1 domain of hepatocyte growth factor has antiangiogenic and antitumor cell effects on hepatocellular carcinoma. Cancer Res. 2008;68(2):404–14. https://doi.org/10.1158/0008-5472.CAN-07-2081.
Diaz-Lezama N, Wu Z, Adan-Castro E, Arnold E, Vazquez-Membrillo M, Arredondo-Zamarripa D, et al. Diabetes enhances the efficacy of AAV2 vectors in the retina: therapeutic effect of AAV2 encoding vasoinhibin and soluble VEGF receptor 1. Lab Investig. 2016;96(3):283–95. https://doi.org/10.1038/labinvest.2015.135.
Ramirez M, Wu Z, Moreno-Carranza B, Jeziorski MC, Arnold E, Diaz-Lezama N, et al. Vasoinhibin gene transfer by adenoassociated virus type 2 protects against VEGF- and diabetes-induced retinal vasopermeability. Invest Ophthalmol Vis Sci. 2011;52(12):8944–50. https://doi.org/10.1167/iovs.11-8190.
Ao H, Liu B, Li H, Lu L. Egr1 mediates retinal vascular dysfunction in diabetes mellitus via promoting p53 transcription. J Cell Mol Med. 2019;23(5):3345–56. https://doi.org/10.1111/jcmm.14225.
Mao XB, Cheng YH, Xu YY. miR-204-5p promotes diabetic retinopathy development via downregulation of microtubule-associated protein 1 light chain 3. Exp Ther Med. 2019;17(4):2945–52. https://doi.org/10.3892/etm.2019.7327.
Dominguez JM 2nd, Hu P, Caballero S, Moldovan L, Verma A, Oudit GY, et al. Adeno-associated virus overexpression of angiotensin-converting Enzyme-2 reverses diabetic retinopathy in type 1 diabetes in mice. Am J Pathol. 2016;186(6):1688–700. https://doi.org/10.1016/j.ajpath.2016.01.023.
Zhang L, Xia H, Han Q, Chen B. Effects of antioxidant gene therapy on the development of diabetic retinopathy and the metabolic memory phenomenon. Graefes Arch Clin Exp Ophthalmol. 2015;253(2):249–59. https://doi.org/10.1007/s00417-014-2827-8.
Xu H, Zhang L, Gu L, Lu L, Gao G, Li W, et al. Subretinal delivery of AAV2-mediated human erythropoietin gene is protective and safe in experimental diabetic retinopathy. Invest Ophthalmol Vis Sci. 2014;55(3):1519–30. https://doi.org/10.1167/iovs.13-13155.
Adhi M, Cashman SM, Kumar-Singh R. Adeno-associated virus mediated delivery of a non-membrane targeted human soluble CD59 attenuates some aspects of diabetic retinopathy in mice. PLoS One. 2013;8(10):e79661. https://doi.org/10.1371/journal.pone.0079661.
Verma A, Shan Z, Lei B, Yuan L, Liu X, Nakagawa T, et al. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Mol Ther. 2012;20(1):28–36. https://doi.org/10.1038/mt.2011.155.
Gong Y, Chang ZP, Ren RT, Wei SH, Zhou HF, Chen XF, et al. Protective effects of adeno-associated virus mediated brain-derived neurotrophic factor expression on retinal ganglion cells in diabetic rats. Cell Mol Neurobiol. 2012;32(3):467–75. https://doi.org/10.1007/s10571-011-9779-x.
Gao N, Wang H, Yin H, Yang Z. Angiotensin II induces calcium-mediated autophagy in podocytes through enhancing reactive oxygen species levels. Chem Biol Interact. 2017;277:110–8. https://doi.org/10.1016/j.cbi.2017.09.010.
Alizadeh S, Mazloom H, Sadeghi A, Emamgholipour S, Golestani A, Noorbakhsh F, et al. Evidence for the link between defective autophagy and inflammation in peripheral blood mononuclear cells of type 2 diabetic patients. J Physiol Biochem. 2018;74(3):369–79. https://doi.org/10.1007/s13105-018-0624-2.
Ghosh P, Sahoo R, Vaidya A, Chorev M, Halperin JA. Role of complement and complement regulatory proteins in the complications of diabetes. Endocr Rev. 2015;36(3):272–88. https://doi.org/10.1210/er.2014-1099.
Mitsuhashi J, Morikawa S, Shimizu K, Ezaki T, Yasuda Y, Hori S. Intravitreal injection of erythropoietin protects against retinal vascular regression at the early stage of diabetic retinopathy in streptozotocin-induced diabetic rats. Exp Eye Res. 2013;106:64–73. https://doi.org/10.1016/j.exer.2012.11.001.
Calderon GD, Juarez OH, Hernandez GE, Punzo SM, De la Cruz ZD. Oxidative stress and diabetic retinopathy: development and treatment. Eye (Lond). 2017;31(8):1122–30. https://doi.org/10.1038/eye.2017.64.
Renna NF, Lembo C, Diez E, Miatello RM. Role of renin-angiotensin system and oxidative stress on vascular inflammation in insulin resistence model. Int J Hypertens. 2013;2013:420979. https://doi.org/10.1155/2013/420979.
Ferreira AJ, Santos RA, Bradford CN, Mecca AP, Sumners C, Katovich MJ, et al. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension. 2010;55(2):207–13. https://doi.org/10.1161/HYPERTENSIONAHA.109.140145.
Huang X, Zhou G, Wu W, Duan Y, Ma G, Song J, et al. Genome editing abrogates angiogenesis in vivo. Nat Commun. 2017;8(1):112. https://doi.org/10.1038/s41467-017-00140-3.
Koo T, Park SW, Jo DH, Kim D, Kim JH, Cho HY, et al. CRISPR-LbCpf1 prevents choroidal neovascularization in a mouse model of age-related macular degeneration. Nat Commun. 2018;9(1):1855. https://doi.org/10.1038/s41467-018-04175-y.
Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9(1):1911. https://doi.org/10.1038/s41467-018-04252-2.
Ciulla TA, Rosenfeld PJ. Anti-vascular endothelial growth factor therapy for neovascular ocular diseases other than age-related macular degeneration. Curr Opin Ophthalmol. 2009;20(3):166–74. https://doi.org/10.1097/ICU.0b013e328329d173.
Garba AO, Mousa SA. Bevasiranib for the treatment of wet, age-related macular degeneration. Ophthalmol Eye Dis. 2010;2:75–83. https://doi.org/10.4137/OED.S4878.
Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, et al. Off-target effects by siRNA can induce toxic phenotype. RNA. 2006;12(7):1188–96. https://doi.org/10.1261/rna.28106.
Rakoczy EP. Gene therapy for the long term treatment of wet AMD. Lancet. 2017;390(10089):6–7. https://doi.org/10.1016/S0140-6736(17)31262-X.
FDA approves hereditary blindness gene therapy. Nat Biotechnol. 2018;36(1):6. https://doi.org/10.1038/nbt0118-6a.
Miller JW, Vandenberghe LH. Breaking and sealing barriers in retinal gene therapy. Mol Ther. 2018;26(9):2081–2. https://doi.org/10.1016/j.ymthe.2018.08.003.
Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5(189):189ra76. https://doi.org/10.1126/scitranslmed.3005708.
Zinn E, Pacouret S, Khaychuk V, Turunen HT, Carvalho LS, Andres-Mateos E, et al. In Silico reconstruction of the viral evolutionary lineage yields a potent gene therapy vector. Cell Rep. 2015;12(6):1056–68. https://doi.org/10.1016/j.celrep.2015.07.019.
McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8(16):1248–54. https://doi.org/10.1038/sj.gt.3301514.
Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15(8):541–55. https://doi.org/10.1038/nrg3763.
Quintino L, Namislo A, Davidsson M, Breger LS, Kavanagh P, Avallone M, et al. Destabilizing domains enable long-term and inert regulation of GDNF expression in the brain. Mol Ther Methods Clin Dev. 2018;11:29–39. https://doi.org/10.1016/j.omtm.2018.08.008.
Datta S, Renwick M, Chau VQ, Zhang F, Nettesheim ER, Lipinski DM, et al. A destabilizing domain allows for fast, noninvasive, conditional control of protein abundance in the mouse eye - implications for ocular gene therapy. Invest Ophthalmol Vis Sci. 2018;59(12):4909–20. https://doi.org/10.1167/iovs.18-24987.
Breger L, Wettergren EE, Quintino L, Lundberg C. Regulated gene therapy. Methods Mol Biol. 2016;1382:57–66. https://doi.org/10.1007/978-1-4939-3271-9_4.
Prentice HM, Biswal MR, Dorey CK, Blanks JC. Hypoxia-regulated retinal glial cell-specific promoter for potential gene therapy in disease. Invest Ophthalmol Vis Sci. 2011;52(12):8562–70. https://doi.org/10.1167/iovs.10-6835.
Geisler A, Fechner H. MicroRNA-regulated viral vectors for gene therapy. World J Exp Med. 2016;6(2):37–54. https://doi.org/10.5493/wjem.v6.i2.37.
Thompson DA, Ali RR, Banin E, Branham KE, Flannery JG, Gamm DM, et al. Advancing therapeutic strategies for inherited retinal degeneration: recommendations from the Monaciano Symposium. Invest Ophthalmol Vis Sci. 2015;56(2):918–31. https://doi.org/10.1167/iovs.14-16049.
Kiser AK, Mladenovich D, Eshraghi F, Bourdeau D, Dagnelie G. Reliability and consistency of visual acuity and contrast sensitivity measures in advanced eye disease. Optom Vis Sci. 2005;82(11):946–54. https://doi.org/10.1097/01.opx.0000187863.12609.7b.
Nair P, Aiello LP, Gardner TW, Jampol LM, Ferris FL III. Report from the NEI/FDA diabetic retinopathy clinical trial design and endpoints workshop. Invest Ophthalmol Vis Sci. 2016;57(13):5127–42. https://doi.org/10.1167/iovs.16-20356.
Ip MS, Zhang J, Ehrlich JS. The clinical importance of changes in diabetic retinopathy severity score. Ophthalmology. 2017;124(5):596–603. https://doi.org/10.1016/j.ophtha.2017.01.003.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council of Australia (GNT1185600) and the Ophthalmic Research Institute of Australia. The Centre for Eye Research Australia receives Operational Infrastructure Support from the Victorian Government.
Author information
Authors and Affiliations
Contributions
Conceptualization—J.H.W., G.S.L. Writing (original draft)—J.H.W., G.S.L. Writing (review and editing)—J.H.W., G.E.R., G.S.L. Funding acquisition—G.S.L.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Immunology, Transplantation, and Regenerative Medicine
Rights and permissions
About this article

Cite this article
Wang, JH., Roberts, G.E. & Liu, GS. Updates on Gene Therapy for Diabetic Retinopathy. Curr Diab Rep 20, 22 (2020). https://doi.org/10.1007/s11892-020-01308-w
Published:
DOI: https://doi.org/10.1007/s11892-020-01308-w