
Abstract
The development of multiple disease-relevant autoantibodies is a hallmark of autoimmune diseases. In autoimmune type 1 diabetes (T1D), a variable time frame of autoimmunity precedes the clinically overt disease. The relevance of T follicular helper (TFH) cells for the immune system is increasingly recognized. Their pivotal contribution to antibody production by providing help to germinal center (GC) B cells facilitates the development of a long-lived humoral immunity. Their complex differentiation process, involving various stages and factors like B cell lymphoma 6 (Bcl6), is strictly controlled, as anomalous regulation of TFH cells is connected with immunopathologies. While the adverse effects of a TFH cell-related insufficient humoral immunity are obvious, the role of increased TFH frequencies in autoimmune diseases like T1D is currently highlighted. High levels of autoantigen trigger an excessive induction of TFH cells, consequently resulting in the production of autoantibodies. Therefore, TFH cells might provide promising approaches for novel therapeutic strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the healthy state, the immune system possesses the critical ability of detecting and eliminating harmful pathogens while at the same time, distinguishing them from the organism’s own tissues. This balance between immunity and tolerance is a complex and tightly regulated process involving different organs and numerous subsets of immune cells. In line with the complexity of the immune system, the heterogeneity of autoimmune diseases like type 1 diabetes (T1D) concerning phenotype and pathology was recently highlighted. These observations emphasize the importance to gain more insight in the underlying mechanisms of autoimmunity and the involved cell types. The recently increased interest in T follicular helper (TFH) cells revealed their important role for the immune system and showed that additional research efforts in the field could contribute to the design of future precision medicines. In this article, we review our current knowledge of TFH cell biology and their considerable role in the context of autoimmune diseases with a particular focus on T1D. Moreover, we discuss the most recent advances in the field, highlighting both their importance as potential novel therapeutic targets and their relevance to exert biomarker function indicative of autoimmune activation state.
Autoimmunity—Immune System out of Balance
Autoimmunity results from an imbalance of the immune system, provoking specific immune responses directed against structures of the self. The concept of autoimmunity was first postulated in the early twentieth century and as one of the pioneers Paul Ehrlich defined an immune-mediated reaction affecting self-constituents as “horror autotoxicus” [1]. There are more than 80 diseases with an autoimmune etiology, most of them with a critically increasing incidence [2]. Because of their high prevalence already in early infancy and their chronic nature, autoimmune diseases are a considerable clinical problem and a significant challenge for the healthcare system.
Autoimmune diseases can be divided in three sequential phases: initiation, propagation, and resolution. The identification of disease-relevant initiation factors is a difficult task because clinical symptoms develop considerably after the incidence of the abnormal immune reaction. However, it is generally accepted in the field that autoimmune diseases originate from a combination of several contributing factors that can be classified as follows: polymorphisms in various genes result in defective regulation or reduced threshold for lymphocyte activation and environmental factors trigger activation of self-reactive lymphocytes that have escaped control [3].
Genome-wide association studies revealed a potential connection between a considerable number of genetic polymorphisms and different autoimmune diseases, for example T1D [4]. Disease-relevant polymorphisms are predominantly located in regulatory regions of genes associated with the immune response and only the accumulation of multiple variations contributes significantly to pathogenesis in single patients [5, 6]. However, some genes like the human leukocyte antigen (HLA) alleles show strong association and have been known for long time [7]. In contrast to most autoimmune diseases which are influenced by numerous genetic polymorphisms, there are two well-studied examples of monogenetic autoimmune diseases. The immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX) and autoimmune polyendocrine syndrome (APS) result directly from an alteration in the FOXP3 and AIRE genes, respectively [8, 9]. Further studies of the autoimmunity-related genes revealed that numerous autoimmunity-connected polymorphisms affect cytokine or cytokine receptor genes, well exemplified by IL-23 receptor which increases proinflammatory capacity of Th17 cells [10] and is polymorphic in various autoimmune diseases [11]. Targeting the underlying pathways with monoclonal antibodies was already subject of clinical trials and showed beneficial effects [12, 13].
While genetic polymorphisms determine the risk of developing autoimmune T1D, the actual initiation of autoimmunity likely requires exogenous triggers such as infections, diet, and other environmental factors [14]. Infections as a potential trigger for autoimmune diseases are under ongoing discussion [15]. While there are numerous examples of postulated associations between infections and autoimmunity like Epstein–Barr virus (EBV) and multiple sclerosis (MS) [16] or periodontal infections and rheumatoid arthritis [17], there are also infections postulated to have a protective effect against autoimmunity for example through antigen-mediated induction of regulatory T cells (Tregs) after an infection with Bacteroides fragilis in a mouse model of MS [18]. In general, a reduced number of infections in developed countries is associated with a higher rate of T1D and MS [19]. Since the microbiome plays an important role for the immune system, its potential to trigger the development of autoimmune diseases by influencing immune reactions is under investigation. For example, there are numerous studies suggesting a connection between microbes and autoimmunity in mice [20–23]. An example for a well-accepted abiotic trigger of autoimmunity is UV radiation, especially in the context of cutaneous lupus. Cell apoptosis caused by UV radiation leads to an excessive supply with self-antigens which can, based on a genetic predisposition, provoke the loss of peripheral tolerance [3, 24].
There are different characteristics of autoimmunity, based on the tolerance mechanism that fails in the specific diseases setting. While in systemic lupus erythematosus (SLE), defects in B cell tolerance seem to play a role [25], other inflammatory autoimmune diseases like T1D are connected with a T cell-dependent autoimmunity [26].
The progression of autoimmunity is associated with increasing inflammation and tissue damage in the affected tissues. This is due to the fact that ongoing tissue damage leads to an accumulation of self-antigens again exacerbating the autoimmune reaction and the coexisting inability to eliminate the self-antigens [3]. Furthermore, the autoimmune reaction as such attracts additional immune cells and induces cytokine secretion, leading to a catastrophic inflammatory loop [3]. With ongoing autoimmunity, the accumulation of effector T cells leads to an imbalance in the ratio of effector T cells to Tregs which, accompanied by Tregs with diminished functionality and effector T cells with a resistance towards suppression, also drives disease progression [27].
A Disease of Increasing Relevance—Characterization and Staging of T1D
As a relevant example for an organ-specific autoimmune disease, T1D is characterized by a breakdown of immunological self-tolerance to insulin-producing islet beta cells and consequently a destruction of these cells by autoreactive T cells [2]. As with autoimmune diseases in general, the incidence of T1D has increased steadily within the last decades [4] accompanied by the critical prediction of a doubling in young children every 20 years [28]. The onset of islet autoimmunity (pre-T1D) is indicated by the appearance of multiple islet autoantibodies against at least two of four well-established autoantigens (insulin, glutamic decarboxylase, insulinoma antigen, and islet zinc transporter) which marks a point of limited return in the pathogenesis of T1D in children [29, 30]. In addition, recently, evidence is emerging that tetraspanin-7 (Tspan-7) can function as an additional autoantigen [31]. However, the time span of the progression to clinical overt T1D is highly variable and can range from a few months to more than two decades, termed as fast versus slow progressing phenotypes [32]. In addition, it was shown that children with slow progression can lose some of their early autoantibodies, first of all insulin autoantibodies. These insights indicate that various mechanisms of immune activation and impaired immune tolerance are involved and can be differentially affected. However, the interplay and contribution of mechanisms that trigger the onset of islet autoimmunity remain incompletely understood.
Like most diseases with an autoimmune etiology, T1D undergoes different stages before the first clinical symptoms occur. Based on specific criteria as genetic polymorphisms and family history, it is possible to predict the risk of developing symptomatic T1D with considerable accuracy [29]. The ability of risk screening and staging of the disease prior to the occurrence of clinical symptoms enables the development of early intervention strategies aiming at the delay of the symptomatic manifestation and eventually its prevention. Based on that, an inclusion of the earliest stages of disease development provides a standardized nomenclature and can therefore be helpful for all phases of prevention approaches from basic research over therapy development and clinical trials to the promotion of precision medicine in clinical practice [30]. The Juvenile Diabetes Research Foundation (JDRF) proposed a refinement of T1D staging in 2015. The main focus of the refinement was the addition of pre-stage 1 which is characterized by genetic susceptibility and genetic risk detection of T1D without islet autoimmunity. The following stage 1 marks the development of two or more T1D-associated islet autoantibodies but with sustained normoglycemia. Here, the 5-year risk of symptomatic disease is approximately 44 %, and the lifetime risk approaches 100 % [32]. In addition to the occurrence of two or more islet autoantibodies, stage 2 is characterized by a loss of functional beta cells and consequently the development of glucose intolerance and disglycemia. The 5-year risk of developing the symptomatic disease increased to 75 %, and the lifetime risk approaches 100 % [33]. Stage 3 marks the occurrence and manifestation of the typical clinical symptoms like polyuria, polydipsia, weight loss, fatigue, diabetic ketoacidosis, and others [30].
The development of autoimmune diseases like T1D is assumed to be based on different concurrent phenomena, among others, the development of islet-specific autoreactive T cells and impairments in Treg-numbers, function, and induction. With respect to the relevant autoreactive lymphocytes, the strong genetic association between the disease and HLA alleles suggests a fundamental involvement of CD4+ T cells [7]. Early studies stated that type 1 helper (Th1) T cells are the predominant drivers of islet destruction in T1D [34]. However, the results of a considerable number of subsequent studies contradicted that dogma [35]. Together with the recognition of additional T cell differentiation states and consequently the blurring of the strict subdivision in Th1 and Th2 subtype, these findings expanded the view to relevant autoreactive T cells and an involvement of for example Th17 cells and TFH cells in disease pathogenesis [36]. The contribution of the latter to T1D and autoimmunity was considerably debated, and there is increasing evidence for an important functional role in that context [37••, 38••].
Characterization of T Follicular Helper Cells
TFH cells are a subset of CD4+ T helper cells specialized to provide help to follicular or GC B cells [39••]. Precursor TFH cells are characterized by abundant expression of C-X-C chemokine receptor type 5 (CXCR5) that is pivotal for their migration to the GCs [39••, 40, 41]. However, CXCR5 expression was shown to be insufficient—and downregulation of C-C chemokine receptor type 7 (CCR7) accompanied by upregulation of programmed cell death protein 1 (PD1) indispensable—for the migration of precursor TFH cells towards the follicular areas in a murine model [42]. It was shown that the vast majority of human peripheral blood CD4+CD45RA−CXCR5+ cells expresses low levels of CCR7 and high levels of PD1 [43]. Thus, TFH cells might more correctly be characterized as activated T cells expressing high levels of CXCR5 and PD1 and low levels of CCR7 [42, 43].
TFH cells represent a discrete cell subset, deviating from other Th cell subsets concerning their surface receptors, intracellular markers, and cytokine profiles. There is evidence, that, despite their diverse origin, human peripheral blood precursor and tonsillar TFH cells as well as murine lymphatic TFH cells are all comprised of a Th1, Th2, and Th17 subtypes and can be discriminated by their cytokine profiles [44–46]. There is an ongoing debate whether TFH cells represent a separate Th cell lineage or a transient activation state [39••]. CXCR3+CCR6− TFH cells with a Th1-like profile mainly secrete interferon-γ (INF-γ), whereas IL-4, IL-5, and IL-13 are secreted by CXCR3−CCR6− TFH cells with Th2-like profile, and IL-17A and IL-22 are secreted by cells with a CXCR3−CCR6+ Th17-like profile [45]. All TFH cell subtypes share the expression of IL-21 with TH17 cells [44, 46].
In humans, both TFH cells with a Th2-like and with a Th17-like profile were found to be capable to induce naïve B cells to secrete immunoglobulins (Ig) via IL-21, whereas TFH cells with a Th1-like profile were unable to provide B cell help. However, the different TFH cell subtypes were shown to variably regulate isotype switching [45].
Thus, TFH cells exhibit an enormous intrinsic heterogeneity enabling them to adapt to various environments, conditions, and needs in order to respond flexibly against any form of pathogen.
Differentiation of T Follicular Helper Cells
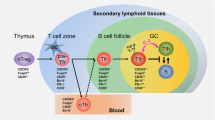
The differentiation of naïve CD4+ T cells into mature TFH cells is a tightly regulated multistep process which is non-linear and involves multiple, alternative differentiation pathways varying between humans and mice (Fig. 1) [39••]. Additionally, TFH cell development depends on the movement of activated T cells out of the T cell zone towards B cell follicles where they interact with cognate B cells and final TFH cell maturation and germinal center formation takes place [47]. Mouse studies showed that TFH cell differentiation begins early in an immune response when naïve CD4+ T cells are primed towards IL-21 producing CXCR5+ICOS+Bcl-6+ early TFH cells by interacting with dendritic cells (DCs) via the T cell antigen-specific receptor (TCR) [37••, 48]. These early TFH cells are further characterized by upregulation of PD1 and downregulation of CCR7 [42] as well as cell adhesion molecule P-selectin glycoprotein ligand 1 (PSGL-1) [49, 50]. E-protein achaete-scute complex homolog 2 (Ascl-2) is a recently identified transcription factor involved in early TFH cell differentiation and might precede expression of the master regulator transcription factor B cell lymphoma 6 protein (Bcl-6). Ascl-2 was identified as an inducer of CXCR5 expression in murine CD4+ T cells in vitro [47]. Bcl-6 expression was shown to be indispensable for initial TFH cell differentiation and found to be transiently upregulated by costimulatory signals of IL-6, IL-21, and inducible T cell costimulator ICOS [51–53]. Recent findings indicated that the latter transiently inactivates Forkhead box protein O1 (Foxo1), which then releases Foxo1-dependent inhibition of Bcl-6 expression and consequently promotes TFH cell differentiation [54•]. The abundance of Foxo1 might be reduced due to increased expression of ICOS, provoked by a loss of Foxp1 or due to ITCH-mediated degradation. Low Foxo1 levels may enhance TFH cell differentiation [55, 56•].

A putative model of TFH cell differentiation and relevant cellular locations. Stages and locations of TFH development, highlighting the complex interplay of multiple genes for TFH cell differentiation, migration, and TFH products (positive regulators shown in blue, negative regulators shown in orange). TFH cells are primed by interacting with dendritic cells (DCs) in the T cell zone. The TFH cell differentiation program is initiated by high expression of Acsl-2; low expression of Id3; and subsequent upregulation of CXCR5, CXCR4, CD69, ICOS, and TCF-7 accompanied by downregulation of CCR7, CCR6, CXCR3, and PSGL-1 and migration of the activated CD4+ T cells towards B cell follicles. At the T cell - B cell border, CD4+CXCR5+ precursor TFH cells interact with cognate B cells and increase Bcl6 expression. This might initiate final TFH cell differentiation in the B cell follicles and germinal center formation. TFH cell differentiation is additionally influenced by MAF, PD1, IL6R, and miRNAs
ICOS additionally triggers the production of IL-21, the major TFH cell cytokine of murine [57] and human [44, 45] TFH cells, and it competes with IL-2 which efficiently inhibits TFH cell differentiation [51, 58]. It is highly expressed on early TFH cells and influences not only TFH cell differentiation but also their migration from the T cell zone towards the border of the B cell follicle via either binding to ICOS-ligand on APCs or via costimulation independent of binding to ICOS-ligand [59]. At the B cell follicle, TFH cells interact with antigen-specific follicular B cells highly expressing ICOS-ligand [42, 49, 50]. These follicular B cells can present antigens enabling division and further maturation of the TFH cells [59–61]. In the GCs, TFH cells continuously interact with APCs triggering their differentiation [36]. The length of this interaction is regulated by signaling lymphocytic activation molecule (SLAM)-associated protein (SAP). Thus, SAP determines the long-term humoral immunity in both humans and mice [62].
Sustained antigenic stimulation maintains the TFH cell phenotype. However, GC TFH cells undergo various changes in their expression of cell surface proteins and transcription factors and in their secretion of molecules [37••].
Both GC TFH cells of murine and human secondary lymphoid organs are characterized as CXCR5highCCR7lowPD1highBcl-6highMafhighSAPhighPSGL-1lowCD200+BTLAhighCCR7low CD4+ T cells that secrete IL-21, IL-4, and C-X-C motif chemokine 13 (CXCL13). Thus, gene expression profiles were found to be highly conserved between species [39••, 63].
TFH cells providing help to GC B cells are capable of exiting the GC and reentering the same or another GC after a certain period of time, showing that they are not terminally differentiated [64]. Additionally, GC TFH cells can differentiate into TFH cells with a memory phenotype [65]. The spontaneous differentiation of T cells from CTLA-4-deficient mice into TFH cells in vivo, accompanied by the formation of large germinal centers, highlighted an important role of CTLA-4 in TFH cell differentiation in mice [66•]. Another factor that plays a considerable role for TFH cell development are microRNAs (miRNAs). These small, 20–22-nucleotide long, noncoding RNAs are critical regulators and modulators of the mammalian immune system [67–69] and are capable to affect complex cellular states [70]. Especially miRNAs that belong to the miR-17∼92 cluster were shown to promote the generation of TFH cells in mice. On contrary, genetic inactivation of miRNA 17∼92 cluster affected TFH cell differentiation, impaired the formation of GCs, and reduced the secretion of high-affinity antibodies. [70]
General Function of TFH Cells
The essential importance of mature TFH cells is due to their crucial contribution to GC development and function. They provide signals to GC B cells that are essential for their survival, differentiation, affinity maturation, isotype switching, and high-affinity antibody secretion. Thus, TFH cells regulate quantity and quality of effector and memory B cell responses and thus the ratio of low-affinity versus high-affinity antibodies. The help signals provided by TFH cells to GC B cells consist of both cytokines and cell-cell interactions and are counterregulated by the duration of the T cell-B cell interaction or inhibitory molecules expressed by other cells and TFH cells themselves [71]. Additionally, follicular regulatory T (TFR) cells which share phenotypic characteristics with TFH cells tightly monitor TFH cells and germinal center B cells by regulating their frequencies. However, research on TFR cells is in its infancy [72]. Besides their critical role for size [73] and cellular composition [74] of the GCs, TFH cells might have further functions outside the GCs which are under ongoing investigation.
TFH Cell Homeostasis—Their Dysregulation Affects Various Diseases
Since TFH cells play a crucial role for generation of high-affinity antibodies, germinal center formation, and development of memory B cells, a close connection between their function and the development and control of numerous diseases is not surprising (Fig. 1). A long-lasting antibody response is the precondition for the protective effect of almost all clinically approved vaccines, and consequently, regular levels of functional TFH cells play an important role for the development of protective immunity, as shown in the context of influenza vaccines [75]. With respect to the control of pathogens, there is a clear correlation between reduced levels of TFH cells and an impaired antibody response. This was exemplified in the context of simian immunodeficiency virus (SIV) in non-human primates [76] and lymphocytic choriomeningitis virus (LCMV) in humans [77]. During a persistent viral infection as in LCMV, prolonged stimulation of the T cell receptor gradually triggers TFH cell instead of Th1 cell response [78]. This might be also true for the infection with dengue virus (DENV), since dengue virus-specific CD4+ T cells show characteristics of TFH cells and are capable to boost antibody production by B cells [79]. Besides their role in the regulation of pathogens, TFH cells are also critically involved in the control of commensal microbiota. Kawanoto et al. 2014 [80] showed that a disturbed gut homeostasis, characterized by unbalanced, less diverse microbiota, results from high TFH cells levels and consequently an enhanced antibody response [80].
TFH deficiencies caused by mutations of TFH-related genes including STAT3, CD40L, and ICOS were shown to be causative for different forms of primary immunodeficiencies [81]. Acquired immunodeficiencies are also accompanied by changes in TFH homeostasis, namely the aggregation of TFH cells in lymph nodes of HIV patients. However, these TFH cells are unable to provide adequate B cell help and thus lead to diminished B cell responses with absent protective antibodies [82]. TFH cells might also be of interest for allergic responses, since exposure to environmental antigens trigger the formation of IL-4 secreting TFH cells identified as Th2 cell precursors and involved in immunoglobulin E (IgE) production in allergic asthma [83].
In autoimmune diseases, TFH cells might contribute to the disease pathology by both excessive generation of autoantibodies and by triggering the formation and maintenance of ectopic follicles. It was shown in a murine model that the amount of antigen determines the quantity and duration of the TFH cell response [37••]. They found a positive correlation between the TFH cell frequency and the number of GC B cells and thus with the humoral immune response provided by these cells. Continuous interaction with B cells and antigen availability were required for the maintenance of TFH cells. However, overstimulation of TFH cells led to a decrease of antigen-specific antibodies and aberrant, pathological levels of autoantibodies that might finally result in autoimmunity. Thus, ongoing antigenic availability as in chronic autoimmune diseases was shown to favor the activation of the TFH cell effector T cell population and the production of autoantibodies [37••].
Two mouse models with a SLE-like phenotype, namely the Roquinsan/san mouse [72] and the BXSB-Yaa mouse [84] showed distinct TFH cell expansion with increased IL-21 production, spontaneous GC formation, and TFH cell accumulation in extrafollicular sites. In contrast, interruption of TFH cell development by blocking ICOS was shown to inhibit TFH cell differentiation, GC formation, and autoantibody production and thus resolved the disease phenotype in the MRL-lpr/lpr (lupus) mouse [49]. The role of TFH cells in human SLE, in which autoantibodies are considered as primarily markers of the disease, is yet not well studied. However, it was shown that elevated frequencies of peripheral blood CD4+CXCR5+ precursor TFH cells were accompanied by increased IL-21 levels and excessive pathogenic autoantibody production in patients with SLE [85]. Equal pathological patterns were observed in other autoimmune diseases such as juvenile dermatomyositis and Sjögren’s syndrome [85]. Increased levels of memory TFH cells and plasma IL-21 were observed in patients with MS compared to healthy controls and patients with complete remission of the disease [86].
In contrast to the beneficial role of Tregs in T1D, TFH cells might play a rather harmful role in this disease. A clear association between IL-21-producing TFH cells and T1D was shown both in a murine model and in human peripheral blood [38••]. The TFH cell signature genes CXCR5, IL-21, PDCD1 (coding for PD1), and Bcl-6 were upregulated in islet-specific T cells of murine pancreatic lymph nodes. Additionally, mRNA levels of CXCR5, ICOS, and IL-21 were significantly elevated in peripheral blood CD4+ T cells from T1D patients compared to those from the control. Another aspect that links T1D with TFH cells is the association with the occurrence of autoantibodies. High-affinity matured autoantibodies were shown to be exclusively generated by the help of TFH cells underlying their indispensable role in T1D [38••].
In line with these findings, a significant increase of circulating TFH cells and elevated levels of IL-21 were shown in patients with T1D compared to healthy controls [87]. It was highlighted that this increase in the percentage of peripheral blood TFH cells is specific to T1D patients with ZnT8 and IA-2, but not to patients with GAD65 serum autoantibodies [88].
In an activated state of the immune system, insulin-producing pancreatic beta cells are the main target of the autoimmune attack causing their progressive destruction. Additionally, autoantibody producing B cells are activated in a T cell-dependent or -independent manner and strengthen the autoimmune reaction. Increased levels of TFH cells might further trigger beta cell destruction by provoking the secretion of islet-specific autoantibodies by plasma cells. On the contrary, Tregs are capable to suppress unwanted autoimmune responses to a certain extent. However, number and function of Tregs were shown to be altered in T1D, and T effector cells might become resistant to Treg suppression over time [26]. Thus, the balance between Tregs, TFH cells, and effector T cells might contribute to the determination of the disease risk [2, 38••, 89].
Recent investigations in autoimmunity highlighted above support the hypothesis that increased TFH cell frequencies and the function of TFH cells contribute to autoimmunity and especially to the pathology of T1D. However, research is in its infancy and knowledge about the function of TFH cells before clinically manifest T1D is limited.
TFH Cell Levels as Novel Markers to Identify Aberrant Immune Activation?
Biomarkers are measurable indicators isolated from serum, urine, or other fluids of the organism that reflect a particular physiological or pathological state. Thus, there is an urgent need to identify relevant biomarkers indicative of variation in immune activation versus regulation and progression to clinically overt disease. As shown above, changes in TFH cell homeostasis have a significant influence on the physiological state of the body (Fig. 2). High levels of functional TFH cells were identified as beneficial and associated with a long-lasting antibody response and protective immunity with regard to infectious diseases [75–78, 81, 90–92]. In contrast, elevated TFH cell levels showed a positive correlation with the presence of allergy [83] and more importantly with the appearance of autoimmune diseases [36]. Abnormal high levels of circulating, peripheral blood CD4+CXCR5+ precursor TFH cells and increased plasma IL-21 levels were observed for various autoimmune diseases such as T1D [37••, 84–87]. The clear impact of TFH cells on the generation of high-affinity matured islet autoantibodies triggering beta cell destruction highlights the clear pathogenic role of TFH cells in T1D. Thus, both high levels of peripheral blood CD4+CXCR5+ precursor TFH cells and IL-21 could be candidates for novel biomarkers relevant for immune activation during islet autoimmunity which warrants further validation. The ratio between Tregs, TFH cells, and effector T cells might determine the disease risk and could serve as an additional biomarker [2, 36, 88]. Furthermore, gene expression analyzes of TFH cell signature genes in peripheral blood CD4+ T cells of healthy people might be an early indicator for the disease risk, since mRNA levels of CXCR5, ICOS, and IL-21 were shown to be significantly elevated in patients with clinically overt T1D [37••]. However, further studies are needed to dissect the disease relevance of these novel T cell-related candidate biomarkers and to precisely characterize murine and human TFH cells to understand the molecular mechanism that regulates their differentiation and functions. Thereby, targeting TFH cells to generate a long-lasting protective antibody response by novel, functional vaccines might be possible in close future [86].

Role and function of TFH cells in immunodeficiency, immune homeostasis, and autoimmunity and levels of potential biomarkers. a The impairment of TFH cell induction or IL-21 signaling (dashed arrows) leads to a decreased antibody production and consequently a limited humoral immunity. b In the healthy state, circulating TFH precursor cells, originating from naive CD4+ T cells, migrate to the germinal center (GC) and differentiate into mature TFH cells. Here, they provide help to B cells, which results in the production of antibodies and the ability to eliminate harmful pathogens. c In autoimmunity, an increased availability of antigens triggers high levels of TFH precursor cells, accompanied by higher levels of serum IL-21 as well as the production of autoantibodies, promoting autoimmunity
Conclusions
Autoimmune diseases like T1D result from a defective regulation of the immune response. Due to their increasing incidence and the complexity of the underlying mechanisms, they are a considerable clinical challenge. The occurrence of autoantibodies is connected with an overreaching TFH cell response, emphasizing this Th cell subset as an important contributor to disease pathogenesis. Since their discovery, more than a decade ago, substantial advances have been made. In addition to their role as a potential target in future preventive and therapeutic strategies, there is increasing evidence to include TFH cell precursor levels, secreted cytokines, and interacting miRNAs, when assessing the disease risk for T1D. However, major knowledge gaps remain especially regarding the complex molecular mechanisms underlying TFH cell differentiation, the function of extrafollicular TFH cells, the interplays between TFH cells and other immune cells as well as the impact of other factors (miRNAs) on TFH cells. Further research will improve our understanding of TFH cell biology and consequently provide approaches for potential therapeutic strategies based on these cells and further clarify their promising role as a biomarker.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ehrlich P, Morgenroth J. Ueber Hämolysine : fünfte Mittheilung. Berliner klinische Wochenschrift. 1901;38:251–7.
Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464(7293):1293–300.
Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. 2015;125(6):2228–33.
Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32(4):457–67.
Zenewicz LA et al. Unraveling the genetics of autoimmunity. Cell. 2010;140(6):791–7.
Marson A, Housley WJ, Hafler DA. Genetic basis of autoimmunity. J Clin Invest. 2015;125(6):2234–41.
Goris A, Liston A. The immunogenetic architecture of autoimmune disease. Cold Spring Harb Perspect Biol. 2012. 4(3).
Bennett CL et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–1.
Finnish-German AC. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet. 1997;17(4):399–403.
Ghoreschi K et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467(7318):967–71.
Vandenbroeck K. Cytokine gene polymorphisms and human autoimmune disease in the era of genome-wide association studies. J Interferon Cytokine Res. 2012;32(4):139–51.
Elliott M et al. Ustekinumab: lessons learned from targeting interleukin-12/23p40 in immune-mediated diseases. Ann N Y Acad Sci. 2009;1182:97–110.
Papp KA et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366(13):1181–9.
Knip M, Simell O. Environmental triggers of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2(7):a007690.
Root-Bernstein R, Fairweather D. Complexities in the relationship between infection and autoimmunity. Curr Allerg Asthma Rep. 2014;14(1):407.
Serafini B et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204(12):2899–912.
Mikuls TR et al. Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthrit Rheumatol. 2014;66(5):1090–100.
Ochoa-Reparaz J et al. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J Immunol. 2010;185(7):4101–8.
Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–41.
Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299(6710):1259–60.
Wills-Karp M, Santeliz J, Karp CL. The germless theory of allergic disease: revisiting the hygiene hypothesis. Nat Rev Immunol. 2001;1(1):69–75.
Abdollahi-Roodsaz S et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118(1):205–16.
Wu HJ et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–27.
Kuhn A, Wenzel J, Weyd H. Photosensitivity, apoptosis, and cytokines in the pathogenesis of lupus erythematosus: a critical review. Clin Rev Allergy Immunol. 2014;47(2):148–62.
Yurasov S et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201(5):703–11.
Bluestone JA, Tang Q, Sedwick CE. T regulatory cells in autoimmune diabetes: past challenges, future prospects. J Clin Immunol. 2008;28(6):677–84.
Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10(12):849–59.
Gale EA. The rise of childhood type 1 diabetes in the 20th century. Diabetes. 2002;51(12):3353–61.
Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity. 2010;32(4):468–78.
Insel RA et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care. 2015;38(10):1964–74.
McLaughlin KA, Richardson CC, Ravishankar A, Brigatti C, Liberati D, Lampasona V, et al. Identification of tetraspanin-7 as a target of autoantibodies in type 1 diabetes. Diabetes. 2016.
Ziegler AG et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA. 2013;309(23):2473–9.
Krischer JP, Type 1 Diabetes TrialNet Study Group . The use of intermediate endpoints in the design of type 1 diabetes prevention trials. Diabetologia. 2013;56(9):1919–24.
Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes. Science. 1995;268(5214):1185–8.
Anderson JT et al. Insulin-dependent diabetes in the NOD mouse model. II. Beta cell destruction in autoimmune diabetes is a TH2 and not a TH1 mediated event. Autoimmunity. 1993;15(2):113–22.
Ferraro A et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes. 2011;60(11):2903–13.
Baumjohann D et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38(3):596–605. This is an important paper showing that the amount of antigen determines the quantity and duration of the TFH cell response in a murine model.
Kenefeck R et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest. 2015;125(1):292–303. This paper highlights a clear association between TFH cells and T1D in a murine model and in human peripheral blood.
Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529–42. This is a useful, broad review of TFH cell differentiation, regulation, function, and their role in disease.
Ansel KM et al. In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med. 1999;190(8):1123–34.
Schaerli P et al. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192(11):1553–62.
Haynes NM et al. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. 2007;179(8):5099–108.
Breitfeld D et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192(11):1545–52.
Chtanova T et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol. 2004;173(1):68–78.
Morita R et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34(1):108–21.
Fazilleau N et al. Follicular helper T cells: lineage and location. Immunity. 2009;30(3):324–35.
Liu X et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature. 2014;507(7493):513–8.
Goenka R et al. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J Immunol. 2011;187(3):1091–5.
Odegard JM et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205(12):2873–86.
Poholek AC et al. In vivo regulation of Bcl6 and T follicular helper cell development. J Immunol. 2010;185(1):313–26.
Choi YS et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34(6):932–46.
Nurieva RI et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325(5943):1001–5.
Johnston RJ et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325(5943):1006–10.
Stone EL et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity. 2015;42(2):239–51. This paper indicates that ICOS transiently inactivates Foxo1 to promote Bcl6 expression and generation of TFH cells.
Xiao N et al. The E3 ubiquitin ligase Itch is required for the differentiation of follicular helper T cells. Nat Immunol. 2014;15(7):657–66.
Wang H et al. The transcription factor Foxp1 is a critical negative regulator of the differentiation of follicular helper T cells. Nat Immunol. 2014;15(7):667–75. This paper demonstrates that CD4+ T cells deficient in Foxp1 might enhance TFH cell differentiation and GC and antibody responses.
Vogelzang A et al. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity. 2008;29(1):127–37.
Johnston RJ et al. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209(2):243–50.
Akiba H et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol. 2005;175(4):2340–8.
Obst R et al. Antigen persistence is required throughout the expansion phase of a CD4(+) T cell response. J Exp Med. 2005;201(10):1555–65.
Weinstein JS et al. B cells in T follicular helper cell development and function: separable roles in delivery of ICOS ligand and antigen. J Immunol. 2014;192(7):3166–79.
Hu J, Havenar-Daughton C, Crotty S. Modulation of SAP dependent T:B cell interactions as a strategy to improve vaccination. Curr Opin Virol. 2013;3(3):363–70.
Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–63.
Shulman Z et al. T follicular helper cell dynamics in germinal centers. Science. 2013;341(6146):673–7.
Kitano M et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity. 2011;34(6):961–72.
Wang CJ et al. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc Natl Acad Sci U S A. 2015;112(2):524–9. Useful paper that shows that CTLA-4 deficiency causes excessive CD28 stimulation and thereby regulation of TFH cell differentiation and GC formation.
Kuipers H et al. Dicer-dependent microRNAs control maturation, function, and maintenance of Langerhans cells in vivo. J Immunol. 2010;185(1):400–9.
Kuipers H, Schnorfeil FM, Brocker T. Differentially expressed microRNAs regulate plasmacytoid vs. conventional dendritic cell development. Mol Immunol. 2010;48(1–3):333–40.
Turner ML, Schnorfeil FM, Brocker T. MicroRNAs regulate dendritic cell differentiation and function. J Immunol. 2011;187(8):3911–7.
Baumjohann D et al. The microRNA cluster miR-17 approximately 92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat Immunol. 2013;14(8):840–8.
Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–57.
Linterman MA et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med. 2009;206(3):561–76.
Hams E et al. Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J Immunol. 2011;186(10):5648–55.
Good-Jacobson KL et al. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. 2010;11(6):535–42.
Bentebibel SE. Induction of ICOS + CXCR3 + CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med. 2013;5(176):176ra32.
Petrovas C et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest. 2012;122(9):3281–94.
Harker JA et al. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science. 2011;334(6057):825–9.
Fahey LM et al. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med. 2011;208(5):987–99.
Rivino L et al. Differential targeting of viral components by CD4+ versus CD8+ T lymphocytes in dengue virus infection. J Virol. 2013;87(5):2693–706.
Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity. 2014;41:152–65.
Al-Herz W et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162.
Cubas RA et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. 2013;19(4):494–9.
Ballesteros-Tato A et al. T follicular helper cell plasticity shapes pathogenic T helper 2 cell-mediated immunity to inhaled house dust mite. Immunity. 2016;44(2):259–73.
Bubier JA et al. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci U S A. 2009;106(5):1518–23.
Szabo K et al. A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjogren’s syndrome and systemic lupus erythematosus. Clin Exp Immunol. 2016;183(1):76–89.
Fan X et al. Circulating CCR7 + ICOS+ memory T follicular helper cells in patients with multiple sclerosis. PLoS One. 2015;10(7):e0134523.
Ferreira RC et al. IL-21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia. 2015;58(4):781–90.
Xu H et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature. 2013;496(7446):523–7.
Yoon JW, Jun HS. Autoimmune destruction of pancreatic beta cells. Am J Ther. 2005;12(6):580–91.
Butler NS et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. 2012;13(2):188–95.
Obeng-Adjei N et al. Circulating Th1-Cell-type Tfh cells that exhibit impaired B cell help are preferentially activated during acute malaria in children. Cell Rep. 2015;13(2):425–39.
Wei M et al. Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat Immunol. 2011;12(3):264–70.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Carolin Daniel (CD) is supported by a Junior Research Group at Helmholtz Zentrum München. CD received support through an associated membership in the CRC1054 of the Deutsche Forschungsgemeinschaft. The work was supported by grants from the Juvenile Diabetes Research Foundation (JDRF 2-SRA-2014-161-Q-R).
Verena B. Ott is supported by Technische Universität München–Institute for Advanced Study, funded by the German Excellence Initiative and the European Union Seventh Framework Programme under grant agreement n° 291763.
Martin G. Scherm is supported by a Helmholtz Diabetes Center Portfolio Grant.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Immunology and Transplantation
Rights and permissions
About this article

Cite this article
Scherm, M.G., Ott, V.B. & Daniel, C. Follicular Helper T Cells in Autoimmunity. Curr Diab Rep 16, 75 (2016). https://doi.org/10.1007/s11892-016-0770-2
Published:
DOI: https://doi.org/10.1007/s11892-016-0770-2