
Abstract
Type 1 diabetes (T1D) is considered a pancreatic beta cell-specific disease that results in absolute insulin deficiency. Nevertheless, clinical studies from 1940 onwards showed that patients with T1D had an abnormal exocrine pancreas due to the presence of subclinical exocrine insufficiency and acinar atrophy. Exocrine abnormalities are an important, and mostly neglected, characteristic associated with T1D. It is however still unclear whether the exocrine dysfunction in T1D is a primary damage caused by the same pathogenic event that led to beta cell destruction or secondary to beta cell loss. In this review, we collect evidence supporting the hypothesis that T1D is a combined endocrine-exocrine disease in which the loss of functional beta cell mass is most clinically apparent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disease in which pancreatic insulin-producing beta cells are destroyed, leading to chronic hyperglycemia. T1D has a variable incidence of 3.9/100,000 to 57.4/100,000, depending on the country, and unfortunately, annual incidence rates are increasing ∼3–4 % worldwide [1]. Abnormalities of the pancreatic exocrine compartment have been described in the past decades in both anatomy and function [2, 3, 4•]. It is unclear whether the exocrine changes in T1D are related to the same genetic, immunological, and environmental events resulting in beta cell destruction or are secondary to the loss of functional β cells. Current conventional opinion supports the notion that insulin acts as a trophic factor for the exocrine compartment [5]. However, in contrast to the well-studied autoimmunity against pancreatic β cells, autoimmune responses towards the exocrine pancreas have garnered less attention. In this review, we will focus on the changes reported in the exocrine pancreas that occur during the natural history of T1D. A novel hypothesis will be presented describing events affecting both exocrine and endocrine compartments that might result in T1D.
Normal Pancreatic Anatomy and Function
The exocrine pancreas unit is composed of acinar, centroacinar, and ductal cells forming the acinus. The exocrine region is divided by connective tissue into lobules containing hundreds of acinar units. Acinar cells secrete more than 20 different enzymes including proteases, lipases, amylases, ribonucleases, and hydrolases into the intralobular ducts, which drain via the main pancreatic duct into the duodenum [6]. These enzymes are stored in zymogen granules as proenzymes that are activated within the duodenum [7].
Exocrine secretion is tightly regulated by the autonomic nervous system in response to a meal and is mediated by numerous hormonal secretagogues and neurotransmitters [8, 9]. One of the most important mediators of exocrine secretion is cholecystokinin (CCK), secreted by I cells in the small intestine and by neurons in the brain [10]. CCK activates sensory afferent neurons in the duodenal mucosa, which in turn stimulates a vago-vagal reflex and the subsequent release of acetylcholine, ultimately leading to exocrine secretion by acinar cells. Exocrine secretory activity is complex and involves many different molecules and activating/inhibitory pathways that are outside of the focus of this review [7, 8, 11].
In contrast to the exocrine portion of the pancreas, only a small proportion of the entire pancreas (1–2 %) is comprised of neuroendocrine cells located in the highly vascularized and innervated islets of Langerhans [6]. Islet neuroendocrine cells are critical for multiple metabolic and physiologic functions of the body. Several hormones, neurotransmitters, and peptides are derived from the islets and these agents could also play important roles in the homeostasis of the exocrine pancreas. Insulin-producing beta cells are the most abundant islet cell type followed by alpha cells (producing glucagon), delta cells (producing somatostatin), gamma cells (producing pancreatic polypeptide), and epsilon cells (producing ghrelin).
Islets are distributed across the acinar tissue facilitating the interaction between endocrine and exocrine tissues. Several groups have postulated that patients with T1D have abnormalities in the exocrine pancreas secondary to the lack of insulin and dysregulation of the endocrine function (reviewed in [12]). Indeed, a direct insulinotropic effect through the local vasculature could result in a high local insulin concentration as proposed by Henderson et al. [13].
Abnormal Pancreatic Anatomy and Function
The most common non-malignant pathologies of the pancreas are acute and chronic pancreatitis. Acute pancreatitis occurs suddenly and may result in life-threatening complications. Chronic pancreatitis results from multiple causes including the progression of acute pancreatitis and can be marked by severe pain and loss of exocrine function. Frequent causes of acute pancreatitis are gallstones secondary to heavy alcohol use. Other causes include direct trauma, high levels of blood fats, high blood calcium, some medications, and certain genetic disorders. An inherited form results in the activation of trypsinogen within the pancreas, leading to autolysis. Genes responsible for this may include trypsin 1, which codes for trypsinogen, SPINK1, which codes for a trypsin inhibitor, or cystic fibrosis transmembrane conductance regulator [14]. A number of infectious agents have been recognized as causes of pancreatitis including viruses (e.g., Coxsackie virus, cytomegalovirus, hepatitis B, herpes simplex virus, mumps, varicella-zoster virus) and bacteria (e.g., Legionella, Leptospira, Mycoplasma, Salmonella, Fungi, Aspergillus) [15]. Conversely, major clinical complications of chronic pancreatitis include malnutrition and weight loss secondary to the loss of acinar cells. In addition, inflammation and injury to pancreatic nerves cause varying degrees of pain [16]. Activation of pancreatic stellate cells leads to fibrosis, and resultant sclerosis could produce pancreatic duct dilation or obstruction [17].
Recent clinical observations lead to the notion that non-endocrine pancreatic disease is a critical factor for the development of diabetes, which is classified as a form of secondary or type 3 diabetes mellitus (T3cDM) by the American Diabetes Association and World Health Organization [18]. Patients with chronic pancreatitis can have high rates of T3cDM (∼78.5 %) while pancreatic cancer seems to be the second common cause (9.8 %) [18]. The incidence of T3cDM caused by exocrine pancreatic disease may comprise 8 % or more of the general diabetic patient population. Diagnosis of T3cDM is often missed and patients are commonly misclassified as type 2 diabetes (T2D) [18]. However, identifying patients with T3cDM is important, since they have special clinical features and require specific therapeutic approaches. In contrast to the management of T1D or T2D, the endocrinopathy in T3cDM is very complex and complicated by additional present comorbidities such as maldigestion and qualitative malnutrition. The failure to correctly diagnose T3cDM leads to failure to implement an appropriate medical therapy of these patients. Easily reversible, important pathological conditions such as exocrine insufficiency, lack of fat-soluble vitamins (especially vitamin D), and impairment of fat hydrolysis and incretin release will not be treated if the physician is not aware of it.
Abnormal Pancreatic Exocrine Function in T1D
The first studies of exocrine pancreatic function in T1D were based on direct CCK and/or secretin stimulation and posterior measurement of enzyme content in duodenal juice [19]. The presence of pancreatic exocrine insufficiency (PEI) was demonstrated in 1943 by Pollard and colleagues [2], who reported a reduction in amylase and lipase after pancreozymin-secretin stimulation that was confirmed afterwards in multiple studies [20–22]. These studies were, however, difficult due to the invasive nature of these test procedures, including tube application into the duodenum and continuous aspiration of duodenal secretion for several hours. Indirect tests became available afterwards and were based mainly on fecal chymotrypsin activity and fecal elastase 1 concentration (FEC) [23–26]. Since these tests did not depend on specialized centers, it was possible to involve larger series of patients in clinical trials. The largest study was carried out as a multi-center trial in Germany, and the prevalence of exocrine pancreatic insufficiency was reported to be 51 % in patients with T1D and 35 % in patients with T2D [24]. The association between exocrine insufficiency and T1D is therefore long, affecting a large proportion of the patients, with an overall very variable prevalence of 43–80 % of PEI in patients with T1D [21, 27]. Recent studies have inversely correlated FEC to diabetes duration (not age) and HbA1c levels while it positively correlated to C-peptide and BMI [28]. It is now evident that exocrine function and morphology should be evaluated in patients with diabetes, at least at the time of diagnosis. Imaging techniques like ultrasound (US), computerized tomography (CT), and magnetic resonance imaging (MRI) can greatly facilitate the assessment of the pancreas and correlate volume with exocrine function [29–31].
Abnormal Pancreatic Exocrine Anatomy and Histology in T1D
The normal human pancreas grows until ∼age 30 with variability in adult pancreas weights or volumes reported (reviewed in [5, 32••, 33]). Smaller pancreata in patients with T1D have been reported by several groups [4•, 5, 34–37]. The autopsy findings were corroborated by radiographic studies using US, CT, and MRI of the pancreas [5, 31, 38–42]. Taken collectively, autopsy weights and clinical imaging studies show pancreatic weights or volumes are reduced by 20–50 % in children and adults with T1D compared to non-diabetic controls.
Potential mechanisms underlying reduced pancreas size in T1D could be due to atrophy, impaired organ growth rate, or a combination of both. Loss of functional beta cell mass and thereby loss of insulinotropic effects on acinar cells has been proposed as a primary mechanism (reviewed in [5, 13]). However, others reported variable and severe acinar atrophy independent of the presence of surviving beta cells [43]. As well, several groups reported no effect of diabetes duration on pancreas weight or size suggesting insulin therapy may not reverse exocrine changes (reviewed in [33]). Finally, a recent report found smaller pancreata in non-diabetic, single autoantibody-positive organ donors [32••]. Larger studies are clearly needed as well as longitudinal imaging studies given the variability in normal pancreas size to better define when pancreas size is reduced during the natural history of T1D.
Exocrine Inflammation in T1D
During the course of T1D, there are evident signs of inflammation possibly as a consequence of exocrine and endocrine tissue loss [44]. The first connection between diabetes and inflammation was reported in 1788 [45] but was defined as pancreatitis only in 1946 by Comfort and colleagues [46]. Foulis et al. noted acute pancreatitis in 6 cases (5 recent onset) in his landmark paper describing the histopathology of insulitis in 95 patients with T1D [47]. Polymorphonuclear cells were present in interstitial tissues and around ducts. Diffuse mononuclear infiltrates were present in 9 patients (4 < 1 year of diabetes onset) to the extent that insulitis was difficult to assess within those regions. Exocrine acini were reported as enlarged with more zymogen granules near insulin-containing islets compared to insulin-negative islets. In contrast, acini from patients with longer duration diabetes (>1 year) tended to be smaller and zymogen depleted (acinar degranulation).
We recently reported that dendritic cells (DCs) and T cells infiltrate the exocrine compartment of cadaveric organ donors with T1D in higher numbers compared to non-diabetic donors [48••]. DCs were also found to be significantly elevated in donors without diabetes but with islet autoantibodies which could have implications for potential antigen presentation and activation of T cells during the initiation of disease. CD4+ T cells were less abundant, but significantly higher numbers were found in the exocrine tissue of T1D donors compared to controls. The presence of DCs correlated with the presence of CD4+ T cells. Moreover, we found high exocrine CD8+ T cell infiltration in T1D donors, independently of disease duration and/or the presence of remaining insulin-producing beta cells. CD8+ T cells have been traditionally implicated in the pathogenesis of T1D, being appointed as the perpetrators of beta cell destruction [49]. Accordingly, cells reactive against beta cell antigens have been found infiltrating the islets of T1D donors [50]. The specificities of CD8+ T cells infiltrating the exocrine compartment remain to be determined as well as the possible consequences for acinar cells and might constitute an interesting new line of investigation.
We also found an unexpected accumulation of neutrophils in the exocrine pancreas of three patients with T1D, but not in the pancreas of T2D and non-diabetic donors [51••]. These pancreas-infiltrating neutrophils mainly localize at the level of very small blood vessels and, to a lesser extent, adjacent to acinar cells in the exocrine tissue. Our finding was also confirmed by another study that reported massive neutrophil infiltration in the exocrine pancreases of two T1D patients who died at disease onset [52]. Intense accumulation of eosinophilic leukocytes was sometimes seen around hyperplastic and hypertrophic islets in infants of mothers with diabetes [53]. Thus, unexpected immune cells (such as those belonging to the innate immune system) can also be found in the pancreas of patients with T1D or exposed to a diabetic environment and are not confined to the beta cells. The presence of B lymphocytes was not investigated in these studies, but the detection of autoantibodies against bile salt-dependent lipase (BSDL), an exocrine pancreatic enzyme specifically expressed by acinar cells, has been previously reported [54]. These antibodies were detected in the serum of 75 % of T1D patients versus 8 % of the controls. Therefore, B cells would most likely be also part of the exocrine inflammatory infiltrate. Additional autoantibodies to pancreas exocrine antigens have been reported in Japanese patients with fulminate T1D and include amylase alpha-2A, carbonic anhydrase II, lactoferrin, and pancreatic cytokeratin [55–57]. However, conflicting studies have been reported regarding levels of carbonic anhydrase II and lactoferrin autoantibodies in Caucasian patients with T1D [58, 59]. Finally, detection of increased levels of complement activation, through its degradation product C4d, has been reported in pancreata from organ donors with T1D [60]. The C4d was found in vascular endothelium and the extracellular matrix of blood vessels and pancreatic ducts. Overall, the presence of immune cells infiltrating the exocrine tissue might have consequences for the acinar cells themselves and contribute to the inflammatory environment of the pancreas, perpetuating the immune destruction of beta cells in T1D patients.
Conclusions
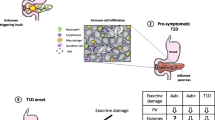
Pancreas exocrine insufficiency is clearly a feature of T1D, and perhaps pre-T1D, but it is generally considered clinically irrelevant compared to beta cell loss. This may have erroneously obscured the role of the exocrine pancreas in the natural history of T1D. However, much more is to be learned about T1D pathogenesis from the perspective of the entire pancreas rather than at solely the islet beta cells (Fig. 1). Despite recent progress, the role of exocrine inflammation in the pathogenesis of T1D remains unclear. An episode of acute pancreatitis or even viral infections might take place in the early stages of the disease, leading to the activation of the immune system and attraction of immune cells to the organ. This inflammation might be then perpetuated and become episodic or chronic in most cases. We believe that there is convincing data to support the hypothesis that T1D is a combined endocrine-exocrine disease in which the loss of functional beta cell mass is most clinically apparent.

Exocrine and endocrine pathways to diabetes. 1-3 T3cDM. A “hit” (e.g., environmental, genetic predisposition) (1) leads to acinar injury that stimulates an inflammatory response (2) in which beta cells are lost as innocent bystanders (3). Destruction of sufficient numbers of beta cells leads to insulin deficiency and T3cDM. 4-7 Classical T1D. A “hit” (e.g., environmental, genetic predisposition) leads to beta cell inflammation, release of self antigens (4), and recruitment of immune cells (5) that target the islet leading to beta cell damage and reduced insulin production (6). Lack of insulin, which is an exocrine growth factor, leads to reduction in exocrine tissue (atrophy) (7). 8-10 Simultaneous T1D-exocrine. A “hit” (e.g., environmental) leads to pancreas inflammation (8), which attracts immune cells (9) to both endocrine and exocrine compartments and induces cellular damage (10). As a consequence, both exocrine insufficiency and insulin deficiency occur which eventually resulting in diabetes onset
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Gale EAM. Epidemiology of type 1 diabetes [internet]. Diapedia. 2014 Aug 13;2104085168 rev. no. 39.
Pollard HM, Miller L, Brewer WA. The external secretion of the pancreas and diabetes mellitus. Am J Dig Dis. 1943;10(1):20–3.
Perczak-Dudkowska B, Niewiedziol B. Exocrine function of the pancreas in juvenile onset diabetes mellitus. I. pH, duodenal content volume and bicarbonate concentration and secretion. Pediatr Pol. 1984;59(8):605–12.
Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14(10):619–33. The first report of decreased pancreatic size in patients with T1D.
Williams AJ, Thrower SL, Sequeiros IM, Ward A, Bickerton AS, Triay JM, et al. Pancreatic volume is reduced in adult patients with recently diagnosed type 1 diabetes. J Clin Endocrinol Metab. 2012;97(11):E2109–13.
Tan GD. The pancreas. Anaesth Intensive Care Med. 2008;9(10):424–7.
Whitcomb DC, Lowe ME. Human pancreatic digestive enzymes. Dig Dis Sci. 2007;52(1):1–17.
Chandra R, Liddle RA. Modulation of pancreatic exocrine and endocrine secretion. Curr Opin Gastroenterol. 2013;29(5):517–22.
Eberhard D, Lammert E. The pancreatic beta-cell in the islet and organ community. Curr Opin Genet Dev. 2009;19(5):469–75.
Pandiri AR. Overview of exocrine pancreatic pathobiology. Toxicol Pathol. 2014;42(1):207–16.
Pandol SJ. The exocrine pancreas. Colloquium series on integrated systems physiology: from molecule to function to disease. San Rafael (CA) 2010.
Piciucchi M, Capurso G, Archibugi L, Delle Fave MM, Capasso M, Delle FG. Exocrine pancreatic insufficiency in diabetic patients: prevalence, mechanisms, and treatment. Int J Endocrinol. 2015;2015:595649.
Henderson JR, Daniel PM, Fraser PA. The pancreas as a single organ: the influence of the endocrine upon the exocrine part of the gland. Gut. 1981;22(2):158–67.
Whitcomb DC. Value of genetic testing in the management of pancreatitis. Gut. 2004;53(11):1710–7.
Parenti DM, Steinberg W, Kang P. Infectious causes of acute pancreatitis. Pancreas. 1996;13(4):356–71.
Di Sebastiano P, di Mola FF, Bockman DE, Friess H, Buchler MW. Chronic pancreatitis: the perspective of pain generation by neuroimmune interaction. Gut. 2003;52(6):907–11.
Lankisch PG, Apte M, Banks PA. Acute pancreatitis. Lancet. 2015.
Ewald N, Kaufmann C, Raspe A, Kloer HU, Bretzel RG, Hardt PD. Prevalence of diabetes mellitus secondary to pancreatic diseases (type 3c). Diabetes Metab Res Rev. 2012;28(4):338–42.
Ewald NH, P.D. Alterations in exocrine pancreatic function in diabetes mellitus. The Pancreapedia. 2015;version 1.0.
Vacca JB, Henke WJ, Knight Jr WA. The exocrine pancreas in diabetes mellitus. Ann Intern Med. 1964;61:242–7.
Lankisch PG, Manthey G, Otto J, Koop H, Talaulicar M, Willms B, et al. Exocrine pancreatic function in insulin-dependent diabetes mellitus. Digestion. 1982;25(3):211–6.
Semakula C, Vandewalle CL, Van Schravendijk CF, Sodoyez JC, Schuit FC, Foriers A, et al. Abnormal circulating pancreatic enzyme activities in more than twenty-five percent of recent-onset insulin-dependent diabetic patients: association of hyperlipasemia with high-titer islet cell antibodies. Belgian Diabetes Registry. Pancreas. 1996;12(4):321–33.
Hardt PD, Krauss A, Bretz L, Porsch-Ozcurumez M, Schnell-Kretschmer H, Maser E, et al. Pancreatic exocrine function in patients with type 1 and type 2 diabetes mellitus. Acta Diabetol. 2000;37(3):105–10.
Hardt PD, Hauenschild A, Nalop J, Marzeion AM, Jaeger C, Teichmann J, et al. High prevalence of exocrine pancreatic insufficiency in diabetes mellitus. A multicenter study screening fecal elastase 1 concentrations in 1,021 diabetic patients. Pancreatol : Off J Int Assoc Pancreatol. 2003;3(5):395–402.
Larger E, Philippe MF, Barbot-Trystram L, Radu A, Rotariu M, Nobecourt E, et al. Pancreatic exocrine function in patients with diabetes. Diabet Med : J Br Diabet Assoc. 2012;29(8):1047–54.
Dominguez-Munoz JE, Hieronymus C, Sauerbruch T, Malfertheiner P. Fecal elastase test: evaluation of a new noninvasive pancreatic function test. Am J Gastroenterol. 1995;90(10):1834–7.
Frier BM, Saunders JH, Wormsley KG, Bouchier IA. Exocrine pancreatic function in juvenile-onset diabetes mellitus. Gut. 1976;17(9):685–91.
Ewald N, Raspe A, Kaufmann C, Bretzel RG, Kloer HU, Hardt PD. Determinants of exocrine pancreatic function as measured by fecal elastase-1 concentrations (FEC) in patients with diabetes mellitus. Eur J Med Res. 2009;14(3):118–22.
Bilgin M, Balci NC, Momtahen AJ, Bilgin Y, Klor HU, Rau WS. MRI and MRCP findings of the pancreas in patients with diabetes mellitus: compared analysis with pancreatic exocrine function determined by fecal elastase 1. J Clin Gastroenterol. 2009;43(2):165–70.
Grippo PJ, Venkatasubramanian PN, Knop RH, Heiferman DM, Iordanescu G, Melstrom LG, et al. Visualization of mouse pancreas architecture using MR microscopy. Am J Pathol. 2011;179(2):610–8.
Sequeiros IM, Hester K, Callaway M, Williams A, Garland Z, Powell T, et al. MRI appearance of the pancreas in patients with cystic fibrosis: a comparison of pancreas volume in diabetic and non-diabetic patients. Br J Radiol. 2010;83(995):921–6.
Campbell-Thompson M, Wasserfall C, Montgomery EL, Atkinson MA, Kaddis JS. Pancreas organ weight in individuals with disease-associated autoantibodies at risk for type 1 diabetes. JAMA. 2012;308(22):2337–9. The first demonstration in humans that pancreas weight is reduced in individuals at risk of developing T1D, thus suggesting that early atrophy of the organ may be an important sub-clinical feature of T1D pathogenesis.
Di Gialleonardo V, de Vries EF, Di Girolamo M, Quintero AM, Dierckx RA, Signore A. Imaging of β-cell mass and insulitis in insulin-dependent (type 1) diabetes mellitus. Endocr Rev. 2012.
Cecil RL. A study of the pathological anatomy of the pancreas in ninety cases of diabetes mellitus. J Exp Med. 1909;11(2):266–90.
MacLean N, Ogilvie RF. Quantitative estimation of the pancreatic islet tissue in diabetic subjects. Diabetes. 1955;4(5):367–76.
Rahier J, Goebbels RM, Henquin JC. Cellular composition of the human diabetic pancreas. Diabetologia. 1983;24(5):366–71.
Foulis AK, Stewart JA. The pancreas in recent-onset type 1 (insulin-dependent) diabetes mellitus: insulin content of islets, insulitis and associated changes in the exocrine acinar tissue. Diabetologia. 1984;26(6):456–61.
Altobelli E, Blasetti A, Verrotti A, Di Giandomenico V, Bonomo L, Chiarelli F. Size of pancreas in children and adolescents with type I (insulin-dependent) diabetes. J Clin Ultrasound. 1998;26(8):391–5.
Fonseca V, Berger LA, Beckett AG, Dandona P. Size of pancreas in diabetes mellitus: a study based on ultrasound. Br Med J (Clin Res Ed). 1985;291(6504):1240–1.
Gilbeau JP, Poncelet V, Libon E, Derue G, Heller FR. The density, contour, and thickness of the pancreas in diabetics: CT findings in 57 patients. AJR Am J Roentgenol. 1992;159(3):527–31.
Goda K, Sasaki E, Nagata K, Fukai M, Ohsawa N, Hahafusa T. Pancreatic volume in type 1 and type 2 diabetes mellitus. Acta Diabetol. 2001;38(3):145–9.
Gaglia JL, Guimaraes AR, Harisinghani M, Turvey SE, Jackson R, Benoist C, et al. Noninvasive imaging of pancreatic islet inflammation in type 1A diabetes patients. J Clin Invest. 2011;121(1):442–5.
Löhr M, Klöppel G. Residual insulin positivity and pancreatic atrophy in relation to duration of chronic type 1 (insulin-dependent) diabetes mellitus and microangiopathy. Diabetologia. 1987;30(10):757–62.
Muniraj T, Aslanian HR, Farrell J, Jamidar PA. Chronic pancreatitis, a comprehensive review and update. Part I: epidemiology, etiology, risk factors, genetics, pathophysiology, and clinical features. Dis Mon : DM. 2014;60(12):530–50.
Beger HG, Buchler M, Kozarek R, Lerch M, Neoptolemos JP, Warshaw A, et al. The pancreas: an integrated textbook of basic science, medicine, and surgery: Wiley; 2 edition; 2008. p. 1024.
Comfort MW, Gambill EE, Baggenstoss AH. Chronic relapsing pancreatitis; a study of 29 cases without associated disease of the biliary or gastrointestinal tract. Gastroenterology. 1946;6:376–408.
Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia. 1986;29(5):267–74.
Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J, von Herrath MG. Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes. 2014;63(11):3880–90. The first demonstration that immune infiltrates in the pancreas of patients with T1D occurs also in the exocrine area.
Willcox A, Richardson S, Bone A, Foulis A, Morgan N. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155(2):173–81.
Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. 2012;209(1):51–60.
Valle A, Giamporcaro GM, Scavini M, Stabilini A, Grogan P, Bianconi E, et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes. 2013;62(6):2072–7. The first demonstration that neutrophils accumulate in the pancreas of patients with T1D especially in the exocrine area.
Korsgren S, Molin Y, Salmela K, Lundgren T, Melhus A, Korsgren O. On the etiology of type 1 diabetes: a new animal model signifying a decisive role for bacteria eliciting an adverse innate immunity response. Am J Pathol. 2012;181(5):1735–48.
Kloppel G. Islet histopathology in diabetes mellitus. Kloppel G, Heitz P, editors. Edinburgh: Churchill Livingstone; 1984. 239 p.
Panicot L, Mas E, Thivolet C, Lombardo D. Circulating antibodies against an exocrine pancreatic enzyme in type 1 diabetes. Diabetes. 1999;48(12):2316–23.
Endo T, Takizawa S, Tanaka S, Takahashi M, Fujii H, Kamisawa T, et al. Amylase alpha-2A autoantibodies: novel marker of autoimmune pancreatitis and fulminant type 1 diabetes. Diabetes. 2009;58(3):732–7.
Taniguchi T, Okazaki K, Okamoto M, Seko S, Tanaka J, Uchida K, et al. High prevalence of autoantibodies against carbonic anhydrase II and lactoferrin in type 1 diabetes: concept of autoimmune exocrinopathy and endocrinopathy of the pancreas. Pancreas. 2003;27(1):26–30.
Kobayashi T, Nakanishi K, Kajio H, Morinaga S, Sugimoto T, Murase T, et al. Pancreatic cytokeratin: an antigen of pancreatic exocrine cell autoantibodies in type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1990;33(6):363–70.
di Cesare E, Previti M, Lombardo F, Mazzù N, di Benedetto A, Cucinotta D. Prevalence of autoantibodies to carbonic anhydrase II and lactoferrin in patients with type 1 diabetes. Ann N Y Acad Sci. 2004;1037:131–2.
Hardt PD, Ewald N, Bröckling K, Tanaka S, Endo T, Kloer HU, et al. Distinct autoantibodies against exocrine pancreatic antigens in European patients with type 1 diabetes mellitus and non-alcoholic chronic pancreatitis. JOP. 2008;9(6):683–9.
Rowe P, Wasserfall C, Croker B, Campbell-Thompson M, Pugliese A, Atkinson M, et al. Increased complement activation in human type 1 diabetes pancreata. Diabetes Care. 2013;36(11):3815–7.
Compliance with Ethics Guidelines
Conflict of Interest
Martha Campbell-Thompson, Teresa Rodriguez-Calvo, and Manuela Battaglia declare they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Pathogenesis of Type 1 Diabetes
Rights and permissions
About this article

Cite this article
Campbell-Thompson, M., Rodriguez-Calvo, T. & Battaglia, M. Abnormalities of the Exocrine Pancreas in Type 1 Diabetes. Curr Diab Rep 15, 79 (2015). https://doi.org/10.1007/s11892-015-0653-y
Published:
DOI: https://doi.org/10.1007/s11892-015-0653-y