
Abstract
Colorectal cancer is one of the major causes of cancer-related mortality in both men and women worldwide. Genetic susceptibility and diet are primary determinants of cancer risk and tumor behavior. Experimental, epidemiological, and clinical data substantiate the beneficial role of n−3 polyunsaturated fatty acids (PUFA) in preventing chronic inflammation and colon cancer. From a mechanistic perspective, n−3 PUFA are pleiotropic and multifaceted with respect to their molecular mechanisms of action. For example, this class of dietary lipid uniquely alters membrane structure/cytoskeletal function, impacting membrane receptor function and downstream signaling cascades, including gene expression profiles and cell phenotype. In addition, n−3 PUFA can synergize with other potential anti-tumor agents, such as fermentable fiber and curcumin. With the rising prevalence of diet-induced obesity, there is also an urgent need to elucidate the link between chronic inflammation in adipose tissue and colon cancer risk in obesity. In this review, we will summarize recent developments linking n−3 PUFA intake, membrane alterations, epigenetic modulation, and effects on obesity-associated colon cancer risk.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Colon cancer is a major public health concern due to the high prevalence of the disease both globally and in the USA [1]. Colon cancer is third in cancer incidence in both men and women and the second leading overall cause of cancer mortality [2]. Epidemiological and migrant population studies have indicated that environmental factors can influence cancer risk [3]. With respect to the focus of this review, diet is considered a key environmental factor impacting colon cancer risk [4, 5]. This is highly relevant because clinical practitioners are currently searching for toxicologically innocuous cancer chemoprevention approaches that are free of safety problems intrinsic to drugs administered over long periods of time. Since a large body of evidence supports the safety and efficacy of dietary or supplemental n−3 polyunsaturated fatty acids (PUFA), e.g., docosahexaenoic acid (DHA, 22:6∆4,7,10,13,16,19) and eicosapentaenoic acid (EPA, 20:5∆5,8,11,14,17), we propose that n−3 PUFA are ideally suited to reduce colon cancer risk. Dietary lipids can alter the cell membrane and tissue fatty acid levels, regulating multiple signaling events, thereby modulating the development of colorectal cancer. It has been shown that tissue fatty acid distribution is related to the incidence of colorectal cancer prognosis [6], with risk increasing by 1–3-fold with respect to n−6 PUFA content. In contrast, risk is reduced by 37–87 % with increasing n−3 PUFA content in colorectal cancerous tissue.
DHA and EPA are enriched in marine fish oil. DHA is a primary structural component of the human brain, cerebral cortex, skin, sperm, testicles, and retina [7]. The parent substrate, α-linolenic acid (18:3n−3; ALA), is found in plant oils, but conversion to EPA and DHA in humans from ALA is low and humans have no other means of synthesizing n−3 PUFAs. Therefore, EPA and DHA can be classified as “essential” nutrients.
Dietary administration of n−3 PUFA in rodent models of colon carcinogenesis has been demonstrated to reduce colon tumor size and multiplicity, compatible with its chemopreventative activity [8–10]. n−3 PUFAs have multiple targets implicated in various stages of cancer development, including cell proliferation, cell survival, angiogenesis, inflammation, metastasis, and epigenetic abnormalities that are crucial to the onset and progression of cancer [11]. In this review, we will focus on three unique aspects of cell signaling modulation by n−3 PUFA: membrane alterations, epigenetic modulation, and impact on obesity-related colon cancer.
n−3 PUFA Effects on Membrane Structure/Signaling
Membrane Lipid Rafts
Cellular membranes are composed of a heterogeneous mixture of lipids and proteins, whose distinct order maintains efficient signal transduction. Membrane lipids can undergo phase separations and interact selectively with membrane proteins and with sub-membrane cytoskeletal elements [12]. Lipid rafts are dynamic and small (10–200 nm) membrane microdomains enriched in sphingolipids and cholesterol, which function as sorting platforms for many membrane-associated proteins [13]. Stabilization of these domains is hypothesized to be maintained by lipid and cytoskeletal influences [14•]. Recent evidence suggests that lipid rafts may modulate the malignant transformation process. For example, the levels of lipid rafts are increased in many types of cancer [15–17]. Additionally, lipid rafts mediate cell signaling events that are often constitutively or hyper-activated in cancer [18–20]. There is also evidence suggesting that disruption of lipid rafts in cancer can lead to increased responsiveness to anti-cancer therapies [21]. Additionally, some anti-cancer drugs have beneficial effects through alteration of the protein content of lipid rafts [22]. In colon cancer, lipid rafts have been shown to function in cell death-mediated signaling [23, 24], cell entry/bioavailability of bioactive compounds [25], and localization of key proteins involved in immune response [26].
Membrane Properties Are Altered by Diet
It has been shown that n−3 PUFA can alter membrane cholesterol and/or sphingomylin content. For example, DHA and EPA reduce cholesterol synthesis in HT29 colon cancer cells [27•]. Fish oil (FO)-fed mice exhibited a ~46 % reduced cholesterol content in colonic caveolae, specialized rafts enriched in the structure protein caveolin-1, which regulate the clustering of signaling proteins such as Ras and eNOS [28]. In addition, a ~40 % reduction in CD4+ T cell lipid raft sphingomyelin levels was observed in mice fed a FO-supplemented diet [29]. Dietary n−3 PUFA are also capable of displacing acylated proteins from lipid raft microdomains in vivo [56] and can alter the size and distribution of cell surface microdomains [30, 41]. n−3 PUFA incorporate into membrane phospholipids, primarily glycerophosphocholine (GPC) and glycerophosphoethanolamine (GPE), in the sn-2 position, creating a highly disordered molecule that is cholesterol-phobic. However, the affinity of DHA-incorporated phospholipids for lipid rafts varies among the different phospholipid subclasses. For example, it has been reported that cholesterol is less soluble in GPE-DHA than in GPC-DHA relative to GPE-oleic acid (OA) and GPC-OA [31–33]. DHA-GPE prefers a non-raft environment, while DHA-GPC prefers a raft environment [34••].
Membrane Lipid Order Modification by n−3 PUFA
Lipid raft stability can be assessed by a variety of polarity-sensitive probes such as Laurdan or di-4-ANEPPDHQ. Quantitative imaging of these probes yields general polarization (GP) values, where higher values reflect a higher membrane order [35, 36]. With respect to diet, membrane order is increased in T cell plasma membranes from FO-fed mice or transgenic mice that produce n−3 PUFA [37, 38••]. Similarly, B cells isolated from mice fed FO show an increase in membrane order in cross-linked cells relative to non-cross-linked cells [39]. This is in contrast to the decrease in membrane order reported in Jurkat cells treated with EPA and DHA [40, 41]. A possible explanation for the differences reported in these studies is that malignant transformed Jurkat cell lines may be inherently different from primary T cells with respect to specific plasma membrane properties. These findings are noteworthy because chronic inflammation increases cancer risk [42, 43]. Specifically, T cell-mediated inflammation in the colon has been linked to the onset of inflammatory bowel diseases (IBD) and to colitis-associated cancer (CAC) [44].
To corroborate the effects of n−3 PUFA on cell surface microdomain organization, immuno-gold electron microscopy of plasma membrane sheets coupled with spatial point analysis of validated microdomain markers has been used [45]. Clustering of probes within cholesterol-dependent (H-Ras) or cholesterol-independent (K-Ras) microdomains exhibits differential sensitivities to PUFA treatment. DHA increases clustering of the lipid raft marker, GFP-tH, and has no effect on the non-raft marker, GFP-tK [30]. This indicates that the effect of DHA is mediated through interactions with lipid raft domains.
n−3 PUFA Displace Signaling Molecules from Raft/Membrane Domains
Many proteins involved in colon cancer cell signaling, including receptors and G proteins, localize to lipid rafts [13]. The epidermal growth factor receptor (EGFR) is a tyrosine kinase that plays a critical role in cell proliferation, survival, and resistance to cancer therapy [46, 47]. EGFR requires lipid raft localization for efficient signaling [48, 49]. n−3 PUFA, in part through a reduction in membrane cholesterol, displaces EGFR from rafts, leading to an altered phosphorylated state [50, 51, 52••]. This in turn suppresses colonocyte downstream signaling events involving EGFR, such as phosphorylation of ERK1/2, STAT3, and Akt and activation of Ras [52••]. A possible explanation for this reduction in signaling may involve an increase in EGFR ubiquitination and internalization.
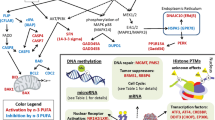
Alteration of raft lipid composition displaces the acylated proteins Lck and LAT from lipid rafts in Jurkat cells treated with EPA [53]. This is associated with the inhibition of T cell signaling by reducing phosphorylation of LAT and phospholipase Cγ1 [54]. A brief summary describing how DHA alters membrane order and EGFR signaling is shown in Fig. 1.

DHA alters membrane order and EGFR signaling. DHA-containing phospholipids incorporate into plasma membrane raft and non-raft domains [34••]. Because of DHA’s low affinity for cholesterol, DHA incorporated into the raft domain reduces cholesterol levels and displaces cholesterol from plasma membrane rafts [28, 31–33]. This reduction of cholesterol displaces EGFR from the raft into the non-raft domain, causing an increase in phosphorylation, and paradoxically decreasing downstream Ras and ERK1/2 signaling, leading to reduced cell proliferation [28, 52••]. DHA also modulates the levels and localization of a critical signaling lipid, phosphoinositide 4,5-bis-phosphate (PIP2) [160••]. This in turn reduces filamentous actin remodeling as well as activation of cytoskeletal regulators, Rac1 and Cdc42, which in turn reduces cell migration [157, 160••]. Cholesterol and the cytoskeleton are major contributors to maintaining membrane order. Interestingly, both these factors are reduced by n−3 PUFA, yet membrane order is increased in cells isolated from FO-fed animals [37, 38••, 39]. This may be attributed to a FO-dependent reduction of two highly disordered fatty acids, arachidonic acid (AA) and n-nervonoylsphingomyelin (C24:1), which could result in a net increase in membrane order [29, 161, 162]
Ras proteins are GTPases, which are targeted to the membrane by farnesylation coupled to either palmitoylation, N, H, and K(A), or a polybasic motif, K(B) [55]. Targeting of palmitoylated N- and H-Ras to the plasma membrane is reduced by DHA, with no effect seen on K(B)-Ras [56]. However, activation of all three isoforms of Ras is attenuated by treatment with DHA [52••]. Collectively, the membrane-altering properties of n−3 PUFA are significant because EGFR and Ras are major drivers of colon cancer [57]. Currently, attempts to directly target Ras have repeatedly failed [58]; therefore, alternate strategies must be pursued. DHA, through modulation of membrane order, may provide a novel therapeutic strategy that may complement current therapies.
Epigenetic Effects of n−3 PUFA
Colon cancer develops through a multistep process that results from the progressive accumulation of mutations and epigenetic alterations in tumor suppressor genes and oncogenes. Epigenetics involves heritable changes in gene expression via posttranslational and posttranscriptional modifications. These modifications typically occur by changes in (i) DNA methylation, (ii) histone modifications, and/or (iii) microRNA (miRNA) expression [59]. These three mechanisms are interconnected to selectively modulate gene expression [60, 61].
-
(i)
In cancer cells, two patterns of DNA methylation can be observed. On the one hand, proto-oncogenes or genes implicated in tumor progression are activated due to global hypomethylation or low levels of methylation. On the other hand, genes such as tumor suppressor genes, implicated in tumor eradication, are silenced due to hypermethylation of their promoter regions [62–64].
-
(ii)
Histone modification by posttranslational processing of their tails directly affects chromatin structure and function and subsequently influences chromatin-based processes, including gene transcription, DNA repair, and DNA replication [65, 66]. The level of histone acetylation is based on the activity of two types of enzymes, namely histone acetyl transferases (HATs) and histone deacetylases/lysine deacetylases (HDACs/KDACs), that regulate the conformation of the chromatin structure to facilitate or hinder the association of DNA repair proteins or transcription factors to chromatin. Hyperacetylation of histones thus can lead to transcriptionally active chromatin. In contrast, deacetylation of histones by HDACs typically leads to a closed (heterochromatin-like) chromatin conformation, thus diminishing accessibility for transcription factors [66, 67]. In this way, HATs serve as activators of gene expression whereas HDACs are typically associated with gene inactivation [68].
-
(iii)
Another modulator of epigenetic modification involves noncoding miRNAs. miRNAs are key regulators of posttranscriptional control of gene expression. miRNAs are aberrantly expressed or mutated in cancer, suggesting that they may function as a novel class of oncogenes or tumor suppressor genes [69–71].
Diet, miRNAs, and Colon Cancer
The effects of colon carcinogen and dietary n−3 PUFA on rodent microRNA expression during the early stages of colon tumorigenesis have been examined [8, 72, 73]. The data indicate that translational alterations are far more extensive relative to transcriptional alterations in mediating malignant transformation. In contrast, transcriptional alterations were found to be more extensive relative to translational alterations in mediating the effects of diet. High-throughput miRNA profiling studies have linked aberrant expression of miRNAs to the development of colon cancer [74, 75]. Specifically, miR-21 is a well-described “oncogenic” miRNA. For example, miR-21 has been positively correlated with colorectal cancer metastasis [76]. Elevated expression of miR-21 has also been reported in colon cancer [8, 77, 78]. miR-21 has anti-apoptotic properties by directly and indirectly targeting several tumor suppressors, PTEN, PDCD4, BCL2, TIMP3, TGFβR2, SPRY3, and RECK [76–86]. The effects of n−3 PUFA on miRNA expression in the gastrointestinal cancers are summarized in Table 1.
Combinatorial Properties of n−3 PUFA with Other Bioactive Agents
It has been demonstrated that dietary FO and fermentable fiber work synergistically to protect against colon carcinogenesis, primarily by enhancing apoptosis [87, 88, 89]. Curcumin, a well-known epigenetic modifier [90–95], with anti-oxidant [96], anti-inflammatory [97], anti-proliferative [98], and anti-angiogenic [99] properties, has also been shown to have synergetic anti-cancer effects when combined with fish oil. For example, we have reported that the combination of fish oil and curcumin can antagonize NFκB activation in the mouse colon following the induction of chronic inflammation [100]. The synergetic effect of DHA and curcumin has also been shown to block insulin-induced colon carcinoma proliferation [101] and inhibit DMBA-induced mammary tumorigenesis in mice [102].
It has also been shown that n−3 PUFA can modulate Wnt signaling, which plays a central role in the physiology and malignant transformation of intestinal stem cells, by suppressing colonocyte nuclear beta-catenin levels [103, 104••, 105, 106, 89]. This is important because perturbations in adult stem cell dynamics are generally believed to represent an early step in colon tumorigenesis [107, 108].
Link Between Obesity and Colon Cancer
Up to 14 and 20 % of all cancer-related deaths may be attributed to obesity in men and women, respectively [109]. Epidemiological data indicate that the risk of colon cancer is strongly associated with increasing body mass index (BMI) [110]. Similar to smoking and a history of colon polyps, a BMI value >25 also significantly increases the risk of colon cancer [111]. In addition to human data, several rodent models have been utilized to demonstrate a link between obesity and increased colon cancer. Studies in mice typically range from 6 to 20 weeks and normally utilize a 32–60 % high-fat lard-based diet resulting in an increased number of colon tumors and aberrant crypt foci as well as increased cell proliferation and reduced apoptosis [112, 113•, 114]. Based on these findings in combination with the rising prevalence of diet-induced obesity, there is an increased need to understand the link between diet, obesity, and colon cancer.
Inflammatory Adipokines
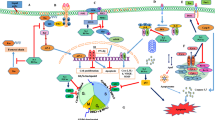
Obesity disrupts the dynamic role of the adipose tissue in energy homeostasis, resulting in the alteration of adipokine signaling and the development of chronic inflammation [115]. Adipose tissue is primarily comprised of mature adipocytes but also contains endothelial cells, adipocyte precursors, fibroblasts, and immune cells. Excess delivery of nutrients to adipose tissue in obesity results in an increase in adipose tissue mass, followed by an increase in immune cell infiltration and thereby an altered production of proinflammatory adipokines (increased IL-1β and leptin, IL-6 and reduced adiponectin), ultimately contributing to the progression of chronic inflammation [116]. M2 macrophages are typically found in the adipose tissue of lean individuals, whereas obese individuals display an increase in M1 macrophages and a shift to a proinflammatory state [117, 118]. Increased infiltration of M1 macrophages in obese mice is also associated with systemic changes in T cell subsets, particularly inflammatory Th1 and Th17 cells [119••]. Studies in both humans and mouse models have provided a clear link between inflammation and cancer [120–122]. The chronic low-grade inflammation associated with obesity may play a key role linking excess adipose tissue, altered adipokine status, and the development of colon cancer by providing a favorable niche for tumor development. Interestingly, diet-induced obese mice that have increased numbers of colonic tumors also have elevated circulating levels of several cytokines and adipokines [113•]. Changes in adipokine status may impact cancer cell growth, as the adipose tissue itself has the ability to secrete several tumor-promoting molecules such as growth factors, proinflammatory cytokines, and adipokines. The most well-characterized adipokines relevant to colon cancer are adiponectin and leptin. These adipokines have been demonstrated to play a role in cell growth, proliferation, apoptosis, angiogenesis, invasion, and migration.
Adiponectin is one of the most abundant protein secreted by adipocytes [123] and can be found in circulation at 2–20 μg/ml [124]. Circulating levels of adiponectin are inversely associated with BMI and visceral adiposity as well as many chronic diseases such as diabetes, cardiovascular disease, and cancer [124–127]. Epidemiological evidence suggests that low adiponectin levels are correlated with the risk of colon cancer [128–130]. Supporting evidence for the protective role of adiponectin against colon cancer has also been demonstrated in mice and cell culture studies. Reported beneficial actions of adiponectin include decreased cell proliferation [131–133], increased apoptosis, reduced number and size of colonies, and decreased adhesion and invasion [133, 134•]. Most impressively, adiponectin appears to reduce the number of polyps, aberrant crypt foci, and tumor size in diet-induced obese mice [132, 134•]. From a mechanistic perspective, adiponectin appears to exert its beneficial actions by promoting phosphorylation of AMPK and LKB1, resulting in changes in cell cycle and inflammatory (p21, p27 cyclin E, STAT3, VEGF, mTOR) pathways [131, 133, 134•].
Leptin is another hormone produced and secreted predominantly by adipocytes, and it is involved in regulating body weight by modifying appetite and energy expenditure [135]. A positive association has been made between leptin and colorectal adenoma in men [130]. Men with the highest leptin concentrations (11–70 ng/ml) had a 3.3-fold increase in risk of colorectal adenoma as compared to those with leptin levels between 1 and 5 ng/ml [136]. In colonic epithelial cells, leptin induces proliferation in a VEGF-dependent manner [137]. In vivo, leptin-deficient mice have reduced colonic tumor size as well as reduced cell proliferation and increased apoptosis [138]. The phosphorylation of STAT3 and upregulation of other inflammatory mediators (IL-6, IL-1β, and CXCL1) appear to be involved in the negative effects of leptin on tumor size [138, 139]. It is also important to note that these two adipokines may interact with each other to influence tumor development [140, 141], an interaction that may be further promoted by obesity.
Inflammatory Cytokines
As mentioned above, the obese adipose tissue becomes infiltrated by immune cells which secrete several inflammatory cytokines, further contributing to the chronic inflammatory environment in obesity. This proinflammatory profile can contribute to the risk of colon cancer. For example, IL-1β has been shown to promote sphere formation and increases in mRNA expression of genes that promote stemness [142, 143]. Cell proliferation and phosphorylation of STAT3 in colon cancer cell lines are induced by IL-6 treatment [131]. Mice deficient in IL-17a display reduced tumor size and number, which is associated with reduced IL-6, IFN-gamma, TNF-alpha, phosphorylated STAT3, and beta-catenin [144].
n−3 PUFA Reduce Obesity-Related Colon Cancer Risk
Since colon cancer development involves adipose-mediated chronic inflammatory processes [145], the adoption of therapeutic strategies to decrease obesity-associated colon tumorigenesis merits consideration. One example of an anti-inflammatory therapeutic is n−3 PUFA. Reductions in the local adipose inflammatory environment have been reported in both humans and rodents treated with n−3 PUFA [119••, 146••]. In mouse models, FO supplementation increased total serum adiponectin levels in diet-induced obese mice and was further enhanced by combination treatment with thiazolidinedione. These changes were associated with a reduction of macrophage infiltration in epididymal white adipose tissue and inflammatory TNF-alpha and MCP1 [147]. Furthermore, fish oil supplemented to a high-fat diet prevented diet-induced obesity, dyslipidemia, hyperinsulinemia, as well as obesity-induced adipocyte hypertrophy and macrophage accumulation in adipose tissue, resulting in increased circulating adiponectin levels [148]. Our laboratory and others have demonstrated that n−3 PUFA are protective against colon tumorigenesis [119••, 149–151] and suppress inflammatory immune cell populations in the colon and adipose tissue [119••, 146••]. Interestingly, in humans, the ratio of n−3/n−6 PUFA was significantly reduced in the visceral white adipose tissue of obese individuals with colorectal cancer and these individuals also exhibited an upregulation of STAT3 and decreased PPAR-gamma and adiponectin levels as compared to normal-weight cancer-free individuals [152••]. The authors were also able to demonstrate that treatment with DHA resulted in a significant reduction in phosphorylated STAT3 and IL-6 in adipocytes from obese colorectal cancer patients [152••].
Conclusion
There is a growing body of experimental, epidemiological, and preclinical evidence indicating that n−3 PUFA, mainly DHA and EPA, are protective against colon tumorigenesis [9, 153–157]. Establishing a causal role of n−3 PUFA in colon cancer prevention would have a major translational impact because these dietary nutrients are safe, well tolerated [158], and relatively inexpensive and provide additional health benefits, such as reduction in mortality [159]. In addition, the ingestion of n−3 PUFA in combination with other agents, such as fermentable fiber and curcumin, may improve their efficacy in colon cancer prevention/therapy. Herein, we have summarized three major mechanisms where n−3 PUFA modulate cancer risk, including membrane lipid order and downstream signaling, epigenetic modulation, and obesity-induced inflammation. Overall, these mechanisms explain some of the actions of an important dietary chemoprotective agent.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90.
Siegel R, Naishadham D, Jemal A. Cancer statistics for Hispanics/Latinos, 2012. CA Cancer J Clin. 2012;62(5):283–98.
Colditz GA, Sellers TA, Trapido E. Epidemiology—identifying the causes and preventability of cancer? Nat Rev Cancer. 2006;6(1):75–83.
Vargas AJ, Thompson PA. Diet and nutrient factors in colorectal cancer risk. Nutr Clin Pract. 2012;27(5):613–23.
Ben Q, Sun Y, Chai R, Qian A, Xu B, Yuan Y. Dietary fiber intake reduces risk for colorectal adenoma: a meta-analysis. Gastroenterology. 2014;146(3):689–99. e6.
Zhang J, Zhang L, Ye X, Chen L, Zhang L, Gao Y, et al. Characteristics of fatty acid distribution is associated with colorectal cancer prognosis. Prostaglandins Leukot Essent Fatty Acids. 2013;88(5):355–60.
Guesnet P, Alessandri JM. Docosahexaenoic acid (DHA) and the developing central nervous system (CNS)—implications for dietary recommendations. Biochimie. 2011;93(1):7–12.
Davidson LA, Wang N, Shah MS, Lupton JR, Ivanov I, Chapkin RS. n-3 Polyunsaturated fatty acids modulate carcinogen-directed non-coding microRNA signatures in rat colon. Carcinogenesis. 2009;30(12):2077–84.
Hull MA. Omega-3 polyunsaturated fatty acids. Best Pract Res Clin Gastroenterol. 2011;25(4–5):547–54.
Cho Y, Turner ND, Davidson LA, Chapkin RS, Carroll RJ, Lupton JR. A chemoprotective fish oil/pectin diet enhances apoptosis via Bcl-2 promoter methylation in rat azoxymethane-induced carcinomas. Exp Biol Med (Maywood). 2012;237(12):1387–93.
Jing K, Wu T, Lim K. Omega-3 polyunsaturated fatty acids and cancer. Anticancer Agents Med Chem. 2013;13(8):1162–77.
Horejsi V, Hrdinka M. Membrane microdomains in immunoreceptor signaling. FEBS Lett. 2014
Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44(4):655–67.
Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta. 2014;1838(2):532–45. The dynamic interaction of membrane lipid rafts and the cytoskeleton are discussed with respect to cell phenotype.
Hazarika P, McCarty MF, Prieto VG, George S, Babu D, Koul D, et al. Up-regulation of Flotillin-2 is associated with melanoma progression and modulates expression of the thrombin receptor protease activated receptor 1. Cancer Res. 2004;64(20):7361–9.
Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168(4):1107–18. quiz 404–5.
Patra SK. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim Biophys Acta. 2008;1785(2):182–206.
Fedida-Metula S, Feldman B, Koshelev V, Levin-Gromiko U, Voronov E, Fishman D. Lipid rafts couple store-operated Ca2+ entry to constitutive activation of PKB/Akt in a Ca2+/calmodulin-, Src- and PP2A-mediated pathway and promote melanoma tumor growth. Carcinogenesis. 2012;33(4):740–50.
Lasserre R, Guo XJ, Conchonaud F, Hamon Y, Hawchar O, Bernard AM, et al. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nat Chem Biol. 2008;4(9):538–47.
Roy UK, Rial NS, Kachel KL, Gerner EW. Activated K-RAS increases polyamine uptake in human colon cancer cells through modulation of caveolar endocytosis. Mol Carcinog. 2008;47(7):538–53.
Irwin ME, Mueller KL, Bohin N, Ge Y, Boerner JL. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J Cell Physiol. 2011;226(9):2316–28.
George KS, Wu S. Lipid raft: a floating island of death or survival. Toxicol Appl Pharmacol. 2012;259(3):311–9.
Lacour S, Hammann A, Grazide S, Lagadic-Gossmann D, Athias A, Sergent O, et al. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 2004;64(10):3593–8.
Rebillard A, Tekpli X, Meurette O, Sergent O, LeMoigne-Muller G, Vernhet L, et al. Cisplatin-induced apoptosis involves membrane fluidification via inhibition of NHE1 in human colon cancer cells. Cancer Res. 2007;67(16):7865–74.
Adachi S, Nagao T, Ingolfsson HI, Maxfield FR, Andersen OS, Kopelovich L, et al. The inhibitory effect of (−)-epigallocatechin gallate on activation of the epidermal growth factor receptor is associated with altered lipid order in HT29 colon cancer cells. Cancer Res. 2007;67(13):6493–501.
Bacso Z, Bene L, Damjanovich L, Damjanovich S. INF-gamma rearranges membrane topography of MHC-I and ICAM-1 in colon carcinoma cells. Biochem Biophys Res Commun. 2002;290(2):635–40.
Gelsomino G, Corsetto PA, Campia I, Montorfano G, Kopecka J, Castella B, et al. Omega 3 fatty acids chemosensitize multidrug resistant colon cancer cells by down-regulating cholesterol synthesis and altering detergent resistant membranes composition. Mol Cancer. 2013;12:137. This cell culture based study provides evidence implicating omega 3 PUFA with lipid rafts and chemosensitivity.
Ma DW, Seo J, Davidson LA, Callaway ES, Fan YY, Lupton JR, et al. n-3 PUFA alter caveolae lipid composition and resident protein localization in mouse colon. FASEB J. 2004;18(9):1040–2.
Fan Y-Y, McMurray DN, Ly LH, Chapkin RS. Dietary (n-3) polyunsaturated fatty acids remodel mouse T-cell lipid rafts. J Nutr. 2003;133(6):1913–20.
Chapkin RS, Wang N, Fan YY, Lupton JR, Prior IA. Docosahexaenoic acid alters the size and distribution of cell surface microdomains. Biochim Biophys Acta. 2008;1778(2):466–71
Shaikh SR, Cherezov V, Caffrey M, Soni SP, LoCascio D, Stillwell W, et al. Molecular organization of cholesterol in unsaturated phosphatidylethanolamines: X-ray diffraction and solid state 2H NMR reveal differences with phosphatidylcholines. J Am Chem Soc. 2006;128(16):5375–83.
Brzustowicz MR, Cherezov V, Caffrey M, Stillwell W, Wassall SR. Molecular organization of cholesterol in polyunsaturated membranes: microdomain formation. Biophys J. 2002;82(1 Pt 1):285–98.
Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69(8):3382–9.
Shaikh SR, Kinnun JJ, Leng X, Williams JA, Wassall SR. How polyunsaturated fatty acids modify molecular organization in membranes: insight from NMR studies of model systems. Biochim Biophys Acta. 2014. The effects of long chain polyunsaturated fatty acids with respect to the biophysical properties of membranes is discussed.
Owen DM, Rentero C, Magenau A, Abu-Siniyeh A, Gaus K. Quantitative imaging of membrane lipid order in cells and organisms. Nat Protoc. 2012;7(1):24–35.
Gaus K, Zech T, Harder T. Visualizing membrane microdomains by Laurdan 2-photon microscopy. Mol Membr Biol. 2006;23(1):41–8.
Kim W, Fan YY, Barhoumi R, Smith R, McMurray DN, Chapkin RS. n-3 polyunsaturated fatty acids suppress the localization and activation of signaling proteins at the immunological synapse in murine CD4+ T cells by affecting lipid raft formation. J Immunol. 2008;181(9):6236–43.
Kim W, Barhoumi R, McMurray DN, Chapkin RS. Dietary fish oil and DHA down-regulate antigen-activated CD4+ T-cells while promoting the formation of liquid-ordered mesodomains. Br J Nutr. 2014;111(2):254–60. Compelling evidence is provided indicating that n-3 PUFA from distinct dietary sources can be integrated into antigen activated CD4+ T-cells, resulting in the modulation of plasma membrane order and translocation of signaling kinases to the immunological synapse.
Rockett BD, Teague H, Harris M, Melton M, Williams J, Wassall SR, et al. Fish oil increases raft size and membrane order of B cells accompanied by differential effects on function. J Lipid Res. 2012;53(4):674–85.
Zech T, Ejsing CS, Gaus K, de Wet B, Shevchenko A, Simons K, et al. Accumulation of raft lipids in T-cell plasma membrane domains engaged in TCR signalling. EMBO J. 2009;28(5):466–76.
Kim W, Khan NA, McMurray DN, Prior IA, Wang N, Chapkin RS. Regulatory activity of polyunsaturated fatty acids in T-cell signaling. Prog Lipid Res. 2010;49(3):250–61.
Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140(6):771–6.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99.
Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J. The soluble interleukin 6 receptor: generation and role in inflammation and cancer. Eur J Cell Biol. 2011;90(6–7):484–94.
Prior IA, Muncke C, Parton RG, Hancock JF. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160(2):165–70.
Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(2 Suppl):21–6.
Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18(2):221–3.
Pike LJ. Growth factor receptors, lipid rafts and caveolae: an evolving story. Biochim Biophys Acta. 2005;1746(3):260–73.
Ringerike T, Blystad FD, Levy FO, Madshus IH, Stang E. Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. J Cell Sci. 2002;115(Pt 6):1331–40.
Rogers KR, Kikawa KD, Mouradian M, Hernandez K, McKinnon KM, Ahwah SM, et al. Docosahexaenoic acid alters epidermal growth factor receptor-related signaling by disrupting its lipid raft association. Carcinogenesis. 2010;31(9):1523–30.
Schley PD, Brindley DN, Field CJ. (n-3) PUFA alter raft lipid composition and decrease epidermal growth factor receptor levels in lipid rafts of human breast cancer cells. J Nutr. 2007;137(3):548–53.
Turk HF, Barhoumi R, Chapkin RS. Alteration of EGFR spatiotemporal dynamics suppresses signal transduction. PLoS One. 2012;7(6):e39682. DHA-induced alteration in both the lateral and subcellular localization of EGFR culminates in the suppression of downstream signal transduction, which has implications for the molecular basis of colon cancer prevention.
Stulnig TM, Huber J, Leitinger N, Imre EM, Angelisova P, Nowotny P, et al. Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. J Biol Chem. 2001;276(40):37335–40.
Zeyda M, Staffler G, Horejsi V, Waldhausl W, Stulnig TM. LAT displacement from lipid rafts as a molecular mechanism for the inhibition of T cell signaling by polyunsaturated fatty acids. J Biol Chem. 2002;277(32):28418–23.
Eisenberg S, Laude AJ, Beckett AJ, Mageean CJ, Aran V, Hernandez-Valladares M, et al. The role of palmitoylation in regulating Ras localization and function. Biochem Soc Trans. 2013;41(1):79–83.
Seo J, Barhoumi R, Johnson AE, Lupton JR, Chapkin RS. Docosahexaenoic acid selectively inhibits plasma membrane targeting of lipidated proteins. FASEB J. 2006;20(6):770–2.
Krasinskas AM. EGFR signaling in colorectal carcinoma. Patholog Res Int. 2011;2011:932932.
Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25(3):272–81.
Sawan C, Vaissiere T, Murr R, Herceg Z. Epigenetic drivers and genetic passengers on the road to cancer. Mutat Res. 2008;642(1–2):1–13.
Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21(1):103–7.
Davis CD, Ross SA. Dietary components impact histone modifications and cancer risk. Nutr Rev. 2007;65(2):88–94.
Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1(2):239–59.
Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56.
Mani S, Herceg Z. DNA demethylating agents and epigenetic therapy of cancer. Adv Genet. 2010;70:327–40.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705.
Sawan C, Herceg Z. Histone modifications and cancer. Adv Genet. 2010;70:57–85.
Steger DJ, Workman JL. Remodeling chromatin structures for transcription: what happens to the histones? Bioessays. 1996;18(11):875–84.
Zhang K, Dent SY. Histone modifying enzymes and cancer: going beyond histones. J Cell Biochem. 2005;96(6):1137–48.
Fabbri M, Calin GA. Epigenetics and miRNAs in human cancer. Adv Genet. 2010;70:87–99.
Vandenboom Ii TG, Li Y, Philip PA, Sarkar FH. MicroRNA and cancer: tiny molecules with major implications. Curr Genomics. 2008;9(2):97–109.
Winter J, Diederichs S. MicroRNA biogenesis and cancer. Methods Mol Biol. 2011;676:3–22.
Davidson LA, Nguyen DV, Hokanson RM, Callaway ES, Isett RB, Turner ND, et al. Chemopreventive n-3 polyunsaturated fatty acids reprogram genetic signatures during colon cancer initiation and progression in the rat. Cancer Res. 2004;64(18):6797–804.
Davidson LA, Wang N, Ivanov I, Goldsby J, Lupton JR, Chapkin RS. Identification of actively translated mRNA transcripts in a rat model of early-stage colon carcinogenesis. Cancer Prev Res (Phila). 2009;2(11):984–94.
Piepoli A, Tavano F, Copetti M, Mazza T, Palumbo O, Panza A, et al. Mirna expression profiles identify drivers in colorectal and pancreatic cancers. PLoS One. 2012;7(3):e33663.
Slaby O, Svoboda M, Michalek J, Vyzula R. MicroRNAs in colorectal cancer: translation of molecular biology into clinical application. Mol Cancer. 2009;8:102.
Slaby O, Svoboda M, Fabian P, Smerdova T, Knoflickova D, Bednarikova M, et al. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology. 2007;72(5–6):397–402.
Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27(15):2128–36.
Wang P, Zou F, Zhang X, Li H, Dulak A, Tomko Jr RJ, et al. microRNA-21 negatively regulates Cdc25A and cell cycle progression in colon cancer cells. Cancer Res. 2009;69(20):8157–65.
Kim YJ, Hwang SJ, Bae YC, Jung JS. MiR-21 regulates adipogenic differentiation through the modulation of TGF-beta signaling in mesenchymal stem cells derived from human adipose tissue. Stem Cells. 2009;27(12):3093–102.
Lu Z, Liu M, Stribinskis V, Klinge CM, Ramos KS, Colburn NH, et al. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene. 2008;27(31):4373–9.
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2):647–58.
Shi L, Chen J, Yang J, Pan T, Zhang S, Wang Z. MiR-21 protected human glioblastoma U87MG cells from chemotherapeutic drug temozolomide induced apoptosis by decreasing Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 2010;1352:255–64.
Song B, Wang C, Liu J, Wang X, Lv L, Wei L, et al. MicroRNA-21 regulates breast cancer invasion partly by targeting tissue inhibitor of metalloproteinase 3 expression. J Exp Clin Cancer Res. 2010;29:29.
Wickramasinghe NS, Manavalan TT, Dougherty SM, Riggs KA, Li Y, Klinge CM. Estradiol downregulates miR-21 expression and increases miR-21 target gene expression in MCF-7 breast cancer cells. Nucleic Acids Res. 2009;37(8):2584–95.
Zhang Z, Li Z, Gao C, Chen P, Chen J, Liu W, et al. miR-21 plays a pivotal role in gastric cancer pathogenesis and progression. Lab Invest. 2008;88(12):1358–66.
Zhou X, Zhang J, Jia Q, Ren Y, Wang Y, Shi L, et al. Reduction of miR-21 induces glioma cell apoptosis via activating caspase 9 and 3. Oncol Rep. 2010;24(1):195–201.
Kolar SS, Barhoumi R, Callaway ES, Fan YY, Wang N, Lupton JR, et al. Synergy between docosahexaenoic acid and butyrate elicits p53-independent apoptosis via mitochondrial Ca(2+) accumulation in colonocytes. Am J Physiol Gastrointest Liver Physiol. 2007;293(5):G935–43.
Kolar SS, Barhoumi R, Lupton JR, Chapkin RS. Docosahexaenoic acid and butyrate synergistically induce colonocyte apoptosis by enhancing mitochondrial Ca2+ accumulation. Cancer Res. 2007;67(11):5561–8.
Vanamala J, Glagolenko A, Yang P, Carroll RJ, Murphy ME, Newman RA, et al. Dietary fish oil and pectin enhance colonocyte apoptosis in part through suppression of PPARdelta/PGE2 and elevation of PGE3. Carcinogenesis. 2008;29(4):790–6.
Chen Y, Shu W, Chen W, Wu Q, Liu H, Cui G. Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch 1 in Raji cells. Basic Clin Pharmacol Toxicol. 2007;101(6):427–33.
Fu S, Kurzrock R. Development of curcumin as an epigenetic agent. Cancer. 2010;116(20):4670–6.
Liu HL, Chen Y, Cui GH, Zhou JF. Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating B-NHL cell line Raji proliferation. Acta Pharmacol Sin. 2005;26(5):603–9.
Reuter S, Gupta SC, Park B, Goel A, Aggarwal BB. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr. 2011;6(2):93–108.
Wu Q, Chen Y, Li X. HDAC1 expression and effect of curcumin on proliferation of Raji cells. J Huazhong Univ Sci Technolog Med Sci. 2006;26(2):199–201. 10.
Ye MX, Li Y, Yin H, Zhang J. Curcumin: updated molecular mechanisms and intervention targets in human lung cancer. Int J Mol Sci. 2012;13(3):3959–78.
Kelkel M, Jacob C, Dicato M, Diederich M. Potential of the dietary antioxidants resveratrol and curcumin in prevention and treatment of hematologic malignancies. Molecules. 2010;15(10):7035–74.
Teiten MH, Eifes S, Reuter S, Duvoix A, Dicato M, Diederich M. Gene expression profiling related to anti-inflammatory properties of curcumin in K562 leukemia cells. Ann N Y Acad Sci. 2009;1171:391–8.
Reuter S, Charlet J, Juncker T, Teiten MH, Dicato M, Diederich M. Effect of curcumin on nuclear factor kappaB signaling pathways in human chronic myelogenous K562 leukemia cells. Ann N Y Acad Sci. 2009;1171:436–47.
Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269(2):199–225.
Jia Q, Ivanov I, Zlatev ZZ, Alaniz RC, Weeks BR, Callaway ES, et al. Dietary fish oil and curcumin combine to modulate colonic cytokinetics and gene expression in dextran sodium sulphate-treated mice. Br J Nutr. 2011;106(4):519–29.
Fenton JI, McCaskey SJ. Curcumin and docosahexaenoic acid block insulin-induced colon carcinoma cell proliferation. Prostaglandins Leukot Essent Fatty Acids. 2013;88(3):219–26.
Siddiqui RA, Harvey KA, Walker C, Altenburg J, Xu Z, Terry C, et al. Characterization of synergistic anti-cancer effects of docosahexaenoic acid and curcumin on DMBA-induced mammary tumorigenesis in mice. BMC Cancer. 2013;13:418.
Bordonaro M, Lazarova DL, Sartorelli AC. Butyrate and Wnt signaling: a possible solution to the puzzle of dietary fiber and colon cancer risk? Cell Cycle. 2008;7(9):1178–83.
Fan YY, Davidson LA, Callaway ES, Goldsby JS, Chapkin RS. Differential effects of 2- and 3-series E-prostaglandins on in vitro expansion of Lgr5+ colonic stem cells. Carcinogenesis. 2014;35(3):606–12. Relative to arachidonic acid-derived PGE2, a known promoter of colon tumorigenesis, eicosapentaenoic acid-derived PGE3 has diminished ability to support colonic stem cell expansion in mouse colonic organoids.
Fujise T, Iwakiri R, Kakimoto T, Shiraishi R, Sakata Y, Wu B, et al. Long-term feeding of various fat diets modulates azoxymethane-induced colon carcinogenesis through Wnt/beta-catenin signaling in rats. Am J Physiol Gastrointest Liver Physiol. 2007;292(4):G1150–6.
Liu Z, Choi SW, Crott JW, Keyes MK, Jang H, Smith DE, et al. Mild depletion of dietary folate combined with other B vitamins alters multiple components of the Wnt pathway in mouse colon. J Nutr. 2007;137(12):2701–8.
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26.
Willis ND, Przyborski SA, Hutchison CJ, Wilson RG. Colonic and colorectal cancer stem cells: progress in the search for putative biomarkers. J Anat. 2008;213(1):59–65.
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–38.
Ning Y, Wang L, Giovannucci EL. A quantitative analysis of body mass index and colorectal cancer: findings from 56 observational studies. Obes Rev. 2010;11(1):19–30.
Ashktorab H, Paydar M, Yazdi S, Namin HH, Sanderson A, Begum R, et al. BMI and the risk of colorectal adenoma in African-Americans. Obesity (Silver Spring). 2014;22(5):1387–91.
Endo H, Hosono K, Fujisawa T, Takahashi H, Sugiyama M, Yoneda K, et al. Involvement of JNK pathway in the promotion of the early stage of colorectal carcinogenesis under high-fat dietary conditions. Gut. 2009;58(12):1637–43.
Olivo-Marston SE, Hursting SD, Perkins SN, Schetter A, Khan M, Croce C, et al. Effects of calorie restriction and diet-induced obesity on murine colon carcinogenesis, growth and inflammatory factors, and microRNA expression. PLoS One. 2014;9(4):e94765. Diet-induced obesity and the suppressive effects of calorie restriction on colon carcinogenesis are associated with alterations in non-coding microRNAs.
Sikalidis AK, Fitch MD, Fleming SE. Diet induced obesity increases the risk of colonic tumorigenesis in mice. Pathol Oncol Res. 2013;19(4):657–66.
Johnson AR, Milner JJ, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. 2012;249(1):218–38.
Karastergiou K, Mohamed-Ali V. The autocrine and paracrine roles of adipokines. Mol Cell Endocrinol. 2010;318(1–2):69–78.
Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–84.
Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56(1):16–23.
Monk JM, Hou TY, Turk HF, Weeks B, Wu C, McMurray DN, et al. Dietary n-3 polyunsaturated fatty acids (PUFA) decrease obesity-associated Th17 cell-mediated inflammation during colitis. PLoS One. 2012;7(11):e49739. n-3 PUFA suppress Th1/Th17 cells and inflammatory macrophage subsets and reconfigure the inflammatory gene expression profile in diverse tissue sites in obese mice following the induction of colitis.
Janakiram NB, Rao CV. The role of inflammation in colon cancer. Adv Exp Med Biol. 2014;816:25–52.
Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–59.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44.
Famulla S, Lamers D, Hartwig S, Passlack W, Horrighs A, Cramer A, et al. Pigment epithelium-derived factor (PEDF) is one of the most abundant proteins secreted by human adipocytes and induces insulin resistance and inflammatory signaling in muscle and fat cells. Int J Obes (Lond). 2011;35(6):762–72.
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257(1):79–83.
DeClercq V, Enns JE, Yeganeh A, Taylor CG, Zahradka P. Modulation of cardiovascular function by adipokines. Cardiovasc Hematol Disord Drug Targets. 2013;13(1):59–72.
Shetty S, Kusminski CM, Scherer PE. Adiponectin in health and disease: evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol Sci. 2009;30(5):234–9.
Yamauchi T, Kadowaki T. Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. Int J Obes (Lond). 2008;32 Suppl 7:S13–8.
Otake S, Takeda H, Fujishima S, Fukui T, Orii T, Sato T, et al. Decreased levels of plasma adiponectin associated with increased risk of colorectal cancer. World J Gastroenterol. 2010;16(10):1252–7.
Wei EK, Giovannucci E, Fuchs CS, Willett WC, Mantzoros CS. Low plasma adiponectin levels and risk of colorectal cancer in men: a prospective study. J Natl Cancer Inst. 2005;97(22):1688–94.
Yamaji T, Iwasaki M, Sasazuki S, Tsugane S. Interaction between adiponectin and leptin influences the risk of colorectal adenoma. Cancer Res. 2010;70(13):5430–7.
Fenton JI, Birmingham JM. Adipokine regulation of colon cancer: adiponectin attenuates interleukin-6-induced colon carcinoma cell proliferation via STAT-3. Mol Carcinog. 2010;49(7):700–9.
Fujisawa T, Endo H, Tomimoto A, Sugiyama M, Takahashi H, Saito S, et al. Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut. 2008;57(11):1531–8.
Kim AY, Lee YS, Kim KH, Lee JH, Lee HK, Jang SH, et al. Adiponectin represses colon cancer cell proliferation via AdipoR1- and -R2-mediated AMPK activation. Mol Endocrinol. 2010;24(7):1441–52.
Moon HS, Liu X, Nagel JM, Chamberland JP, Diakopoulos KN, Brinkoetter MT, et al. Salutary effects of adiponectin on colon cancer: in vivo and in vitro studies in mice. Gut. 2013;62(4):561–70. Adipose-derived adipokines are directly implicated in colon cancer risk.
Pan H, Guo J, Su Z. Advances in understanding the interrelations between leptin resistance and obesity. Physiol Behav. 2014;130:157–69.
Chia VM, Newcomb PA, Lampe JW, White E, Mandelson MT, McTiernan A, et al. Leptin concentrations, leptin receptor polymorphisms, and colorectal adenoma risk. Cancer Epidemiol Biomarkers Prev. 2007;16(12):2697–703.
Birmingham JM, Busik JV, Hansen-Smith FM, Fenton JI. Novel mechanism for obesity-induced colon cancer progression. Carcinogenesis. 2009;30(4):690–7.
Endo H, Hosono K, Uchiyama T, Sakai E, Sugiyama M, Takahashi H, et al. Leptin acts as a growth factor for colorectal tumours at stages subsequent to tumour initiation in murine colon carcinogenesis. Gut. 2011;60(10):1363–71.
Padidar S, Farquharson AJ, Williams LM, Kelaiditi E, Hoggard N, Arthur JR, et al. Leptin up-regulates pro-inflammatory cytokines in discrete cells within mouse colon. J Cell Physiol. 2011;226(8):2123–30.
Yamaji T, Iwasaki M, Sasazuki S, Tsugane S. Interaction between adiponectin and leptin influences the risk of colorectal adenoma. Cancer Res. 2010;70(13):5430–7.
Fenton JI, Birmingham JM, Hursting SD, Hord NG. Adiponectin blocks multiple signaling cascades associated with leptin-induced cell proliferation in Apc Min/+colon epithelial cells. Int J Cancer. 2008;122(11):2437–45.
Li Y, Wang L, Pappan L, Galliher-Beckley A, Shi J. IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol Cancer. 2012;11:87.
Wang L, Liu Z, Li Y, Pappan L, Galliher-Beckley A, Shi J. Pro-inflammatory cytokine interleukin-1beta promotes the development of intestinal stem cells. Inflamm Res. 2012;61(10):1085–92.
Hyun YS, Han DS, Lee AR, Eun CS, Youn J, Kim HY. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis. 2012;33(4):931–6.
Hillenbrand A, Fassler J, Huber N, Xu P, Henne-Bruns D, Templin M et al. Changed adipocytokine concentrations in colorectal tumor patients and morbidly obese patients compared to healthy controls. BMC cancer. 2012;12:545–2407–12–545.
Itariu BK, Zeyda M, Hochbrugger EE, Neuhofer A, Prager G, Schindler K, et al. Long-chain n-3 PUFAs reduce adipose tissue and systemic inflammation in severely obese nondiabetic patients: a randomized controlled trial. Am JClin Nutr. 2012;96(5):1137–49. Treatment with long-chain n-3 PUFAs favorably modulated adipose tissue and systemic inflammation in severely obese non-diabetic patients and improved lipid metabolism.
Jilkova ZM, Hensler M, Medrikova D, Janovska P, Horakova O, Rossmeisl M, et al. Adipose tissue-related proteins locally associated with resolution of inflammation in obese mice. Int J Obes (Lond). 2014;38(2):216–23.
Kuda O, Jelenik T, Jilkova Z, Flachs P, Rossmeisl M, Hensler M, et al. n-3 fatty acids and rosiglitazone improve insulin sensitivity through additive stimulatory effects on muscle glycogen synthesis in mice fed a high-fat diet. Diabetologia. 2009;52(5):941–51.
Jia Q, Lupton JR, Smith R, Weeks BR, Callaway E, Davidson LA, et al. Reduced colitis-associated colon cancer in fat-1 (n-3 fatty acid desaturase) transgenic mice. Cancer Res. 2008;68(10):3985–91.
Nowak J, Weylandt KH, Habbel P, Wang J, Dignass A, Glickman JN, et al. Colitis-associated colon tumorigenesis is suppressed in transgenic mice rich in endogenous n-3 fatty acids. Carcinogenesis. 2007;28(9):1991–5.
Rao CV, Hirose Y, Indranie C, Reddy BS. Modulation of experimental colon tumorigenesis by types and amounts of dietary fatty acids. Cancer Res. 2001;61(5):1927–33.
D’Archivio M, Scazzocchio B, Giammarioli S, Fiani ML, Vari R, Santangelo C, et al. Omega 3-PUFAs exert anti-inflammatory activity in visceral adipocytes from colorectal cancer patients. PLoS One. 2013;8(10):e77432. Adipocyte dysfunction occurs in colon cancer patients creating a pro-inflammatory environment that might influence cancer development. The protective potential of DHA in re-establishing the equilibrium between pro- and anti-inflammatory factors is documented.
Chapkin RS, McMurray DN, Lupton JR. Colon cancer, fatty acids and anti-inflammatory compounds. Curr Opin Gastroenterol. 2007;23(1):48–54.
Cheng J, Ogawa K, Kuriki K, Yokoyama Y, Kamiya T, Seno K, et al. Increased intake of n-3 polyunsaturated fatty acids elevates the level of apoptosis in the normal sigmoid colon of patients polypectomized for adenomas/tumors. Cancer Lett. 2003;193(1):17–24.
Cockbain AJ. Omega-3 polyunsaturated fatty acids for the treatment and prevention of colorectal cancer. Gut. 2012;61(1):135–49.
Courtney ED. Eicosapentaenoic acid (EPA) reduces crypt cell proliferation and increases apoptosis in normal colonic mucosa in subjects with a history of colorectal adenomas. Int J Color Dis. 2007;22(7):765–76.
Turk HF, Monk JM, Fan YY, Callaway ES, Weeks B, Chapkin RS. Inhibitory effects of omega-3 fatty acids on injury induced epidermal growth factor transactivation contribute to delayed wound healing. Am J Physiol Cell Physiol. 2013;304(9):C905–17.
Lien EL. Toxicology and safety of DHA. Prostaglandins Leukot Essent Fatty Acids. 2009;81(2–3):125–32.
Bell GA, Kantor ED, Lampe JW, Kristal AR, Heckbert SR, White E. Intake of long-chain omega-3 fatty acids from diet and supplements in relation to mortality. Am J Epidemiol. 2014;179(6):710–20.
Hou TY, Monk JM, Fan YY, Barhoumi R, Chen YQ, Rivera GM, et al. n-3 polyunsaturated fatty acids suppress phosphatidylinositol 4,5-bisphosphate-dependent actin remodelling during CD4+ T-cell activation. Biochem J. 2012;443(1):27–37. Cogent evidence demonstrates for the first time that DHA modulates PI(4,5)P2-dependent actin remodeling by decreasing steady-state PI(4,5)P2 levels. These findings highlight a novel modality by which n-3 PUFA influence membrane organization, thereby modulating biological responses.
Mate S, Busto JV, Garcia-Arribas AB, Sot J, Vazquez R, Herlax V, et al. N-Nervonoylsphingomyelin (c24:1) prevents lateral heterogeneity in cholesterol-containing membranes. Biophys J. 2014;106(12):2606–16.
Rajamoorthi K, Petrache HI, McIntosh TJ, Brown MF. Packing and viscoelasticity of polyunsaturated omega-3 and omega-6 lipid bilayers as seen by (2)H NMR and X-ray diffraction. J Am Chem Soc. 2005;127(5):1576–88.
Shah MS, Schwartz SL, Zhao C, Davidson LA, Zhou B, Lupton JR, et al. Integrated microRNA and mRNA expression profiling in a rat colon carcinogenesis model: effect of a chemo-protective diet. Physiol Genomics. 2011;43(10):640–54.
Gil-Zamorano J, Martin R, Daimiel L, Richardson K, Giordano E, Nicod N, et al. Docosahexaenoic acid modulates the enterocyte Caco-2 cell expression of microRNAs involved in lipid metabolism. J Nutr. 2014;144(5):575–85.
Sun H, Meng X, Han J, Zhang Z, Wang B, Bai X, et al. Anti-cancer activity of DHA on gastric cancer—an in vitro and in vivo study. Tumour Biol. 2013;34(6):3791–800.
Acknowledgments
This work was supported by the American Institute for Cancer Research (AICR); Cancer Prevention and Research Institute of Texas (CPRIT); National Institutes of Health grants CA129444, CA168312, and P30ES023512; and USDA–NIFA Grant Designing Foods for Health 2010-34402-20875. The authors would also like to thank Tim Hou for formatting assistance.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Robert S. Chapkin, Vanessa DeClercq, Eunjoo Kim, Natividad Roberto Fuentes, and Yang-Yi Fan declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chapkin, R.S., DeClercq, V., Kim, E. et al. Mechanisms by Which Pleiotropic Amphiphilic n−3 PUFA Reduce Colon Cancer Risk. Curr Colorectal Cancer Rep 10, 442–452 (2014). https://doi.org/10.1007/s11888-014-0241-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11888-014-0241-6