
Abstract
Purpose of Review
This review aims to describe the latest advances in autonomic neuromodulation approaches to treating cardiac arrhythmias, with a focus on ventricular arrhythmias.
Recent Findings
The increasing understanding of neuronal remodeling in cardiac diseases has led to the development and improvement of novel neuromodulation therapies targeting multiple levels of the autonomic nervous system. Thoracic epidural anesthesia, spinal cord stimulation, stellate ganglion modulatory therapies, vagal stimulation, renal denervation, and interventions on the intracardiac nervous system have all been studied in preclinical models, with encouraging preliminary clinical data.
Summary
The autonomic nervous system regulates all the electrical processes of the heart and plays an important role in the pathophysiology of cardiac arrhythmias. Despite recent advances in the clinical application of cardiac neuromodulation, our comprehension of the anatomy and function of the cardiac autonomic nervous system is still limited. Hopefully in the near future, more preclinical data combined with larger clinical trials will lead to further improvements in neuromodulatory treatment for heart rhythm disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The heart is richly innervated by afferent as well as sympathetic and parasympathetic efferent nerves. Their activity is embodied in a complex series of peripheral and central feedback loops that enable autonomic control of cardiac function. The entire autonomic nervous system (ANS) aims to maintain cardiac homeostasis in the face of environmental perturbations by ensuring adequate and timely responses to stimuli. Nevertheless, in pathological conditions such as myocardial infarction/ischemia and heart failure, autonomic reflexes may become detrimental and lead to life-threatening arrhythmias and progression of cardiac dysfunction. In this review, we will first discuss the basic anatomy, physiology, and pathophysiology of the cardiac ANS, with particular focus on its role in the genesis of ventricular arrhythmias (VAs). Then, we will provide an overview about the main clinical applications of ANS modulation in the treatment of VAs.
Functional Anatomy of the Cardiac Neuraxis
The cardiac neuraxis is divided into the intrinsic and extracardiac systems. The intrinsic cardiac nervous system (ICNS) is a network of ganglia and interconnecting nerves located in multiple ganglionated plexi (GPs) in the epicardial fat pads [1]. The extracardiac system includes the mediastinal plexus, cervico-thoracic ganglia, nodose ganglia, dorsal root ganglia (DRG), spinal cord, and the brainstem.
Efferent sympathetic preganglionic neurons have their soma in the intermediolateral column [2] of spinal cord and synapse on postganglionic neurons located in the first four ganglia of the sympathetic paravertebral chain (from T1 to T4, T1 being the lower half/third of the stellate ganglion).
The axons arising from these ganglia reach the heart through the cardiopulmonary nerves. In addition, sympathetic postganglionic neurons projecting to the heart are also localized in each major intrinsic cardiac GP [3, 4].
The soma of efferent parasympathetic preganglionic neurons is mainly located in the ventral lateral region of the nucleus ambiguus, giving rise to the vagus nerve in the periphery. The vagal trunk is a mixed nerve, composed of about 80% A∂ and C-type afferent fibers, and about 20% group B efferent fibers. The cardiomotor fibers project onto parasympathetic ganglia of the ICNS. The cardiac vagal branches, together with the cardiopulmonary nerves, constitute the mediastinal plexus.
Finally, the neuronal bodies of cardiac afferent neurons are contained in the nodose ganglia, the DRG from C7-T4 spinal cord [5], as well as intrinsic cardiac ganglia [6], providing the anatomical bases for a multiple-loop neuronal control.
The ICNS is also referred to as the “little brain” on the heart [7], because it is the first, independent, station of the cardiac ANS. It allows for local, beat-to-beat, reflex control of all heart functions and responses as well as integration and coordination with higher centers for middle- and long-term reflexes. This is accomplished through the presence of afferent sensory neurons, efferent motor neurons, local circuit neurons (neurons that project axons only to adjacent neurons), and interneurons (neurons that project axons to other ganglia). The majority of efferent neurons in the ICNS is cholinergic and responds to preganglionic control from efferent fibers in the vagus nerve. Cholinergic and adrenergic neurons are contained in each atrial and ventricular GP and control divergent regions of the heart [8]. The highest density of ICNS neurons in humans is located on the posterior surface of the atria.
Cardiac ANS Impact on Arrhythmogenesis
Sympathetic-Parasympathetic Effects at the Cardiac Level
The most important consequence of sympathetic neuronal activation is the net increase in norepinephrine (NE) and other co-transmitter (e.g., neuropeptide Y) release at the cardiac level. The spatially and temporally heterogeneous neuronal release of NE has been postulated to result in increased arrhythmia susceptibility. NE promotes arrhythmogenesis at the ventricular level by enhancing triggered activity (both early and delayed after depolarization) [9], automaticity and reentry [10], then finally leading to an increase in ventricular tachycardia burden combined with a decrease in ventricular fibrillation threshold [11, 12]. Accordingly, it was recently shown in a porcine model [13] that sympathetic nerve stimulation, and not circulating norepinephrine, modulates T-peak to T-end interval (an ECG marker of dispersion of repolarization) by increasing global dispersion of repolarization. All these effects—particularly the increased dispersion in ventricular repolarization—are further exacerbated in the setting of structural heart diseases (SHDs), where denervation and reinnervation (nerve sprouting) occur.
The ratio between parasympathetic and sympathetic fibers at the cardiac level conveys important functional implications. Indeed, the two systems mutually antagonize one another, both at the presynaptic and postsynaptic levels. Acetylcholine (ACh) inhibits NE release from sympathetic synapses [14] and antagonizes NE effects at cellular levels. The co-trasmitters neuropeptide Y and galanin, released from sympathetic nerve terminals during high levels of sympathetic drive, can reduce Ach release from parasympathetic terminals [15]. Finally, ACh also exerts direct anti-inflammatory and anti-apoptotic effects at cardiac level through purinergic pathways [16].
Effects of Enhanced Cardiac Afferent Signaling
One pathophysiological basis of acute and chronic cardiac sympathoexcitation is the overactivation of powerful autonomic reflexes. The afferent branch of these reflexes is provided by extracardiac and intracardiac receptors. In response to reduced cardiac output, baroreceptors located in the carotid sinus and renal arteries trigger increased sympathetic outflow and decreased parasympathetic activity, as well as hyperactivation of the renin-angiotensin aldosterone system [17]. At the cardiac level, the pathological activation of afferent neurons leads to a powerful increase in sympathetic efferent drive to the heart [18]. The immediate consequence of this reflex, also known as cardiac sympathetic afferent reflex (CSAR), is a strong inotropic and chronotropic response, aimed to sustain cardiac output. The inevitable adverse effect is a consistent increase in metabolic demand and in arrhythmic susceptibility. Chronic activation of the CSAR is also associated with parasympathetic withdrawal at the central level, which strongly enhances arrhythmogenesis [19]. From a molecular point of view, the CSAR is largely mediated by the vanilloid receptor 1 (VR1) or transient receptor potential vanilloid 1 (TRPV1) channel [20]. TRPV1 channels are expressed in both unmyelinated C-fibers and thinly myelinated Aδ- fibers and facilitate nociception during myocardial ischemia [21]. These channels are able to transduce multiple stimuli, including noxious heat, bradykinin [22], acidity, and free radicals [23]. TRPV1-mediated CSAR activation was shown to be upregulated in congestive heart failure (CHF) animal models [24]. Thus, these channels are emerging as a potential therapeutic target in cardiac diseases marked by exaggerated sympathoexcitation. Selective chemical ablation of TPRV1-expressing afferents by epicardial application of resiniferatoxin (a toxic activator of the VR1 channel) provided powerful protective effects against adverse cardiac remodeling and autonomic dysfunction induced by myocardial infarction (MI) in rats [25]. From a clinical standpoint, the protective effect of pre-infarction angina has been attributed to ischemic preconditioning and to better collateral circulation development. Yet, a blunted acute activation of the CSAR in response to the ischemic trigger might be involved as well. Altogether, these data suggest that the afferent component of CSAR is a very appealing therapeutic target, although much more data are needed to fully unravel its physiological role. The possibility of achieving a transient, reversible block of cardiac afferent fibers to acutely counteract exaggerated sympathoexcitation (such as during acute myocardial ischemia and/or refractory electrical storms) is still unexplored, but could be extremely beneficial.
Intra- and Extra-Cardiac Neural Remodeling
The process of cardiac neuronal remodeling was mainly studied in ischemic models, but also occurs within other forms of cardiomyopathies (CM) [26]. It includes denervation as well as nerve sprouting (reinnervation). The denervated myocardium is characterized by an increased susceptibility to catecholamines, also known as denervation supersensitivity [27], while the reinnervated myocardium is characterized by a constitutively abnormal afferent signaling combined with a very patchy distribution of sympathetic efferent fibers. After MI, denervation occurs not only within the dense scar, but also in the viable myocardium surrounding the scar and in remote regions [28]. Clinical data confirm that the viable myocardium surrounding the scar is characterized by a combination of blunted response to reflex sympathetic activation and hypersensitivity to circulating catecholamine [29], both of which contribute to a functional increase in ventricular repolarization dispersion. Accordingly, successful ablation procedures for monomorphic ventricular tachycardia (VT) after MI most frequently target the border zone regions [30] defined through high-density voltage maps. Yet, neuronal efferent fibers are more sensitive to ischemia than cardiomyocytes and efferent denervation may also occur in regions surrounding the scar with normal voltages.
A relatively novel method which draws on our understanding of cardiac neuronal remodeling to aid in arrhythmia treatment is the use of scintigraphic techniques such as I-123 metaiodobenzylguanidine (123I-mIBG) to quantify sympathetic denervation in vivo in humans. 123I-mIBG imaging can also be used to create 3-dimensional innervation models and register them into high-density voltage maps to guide VT ablation procedures. A recent human study [31•] confirmed that 123I-mIBG innervation defects are larger than bipolar voltage–defined scar and cannot be detected with standard voltage criteria. In this study, as much as 36 % of successful VT ablation sites had normal voltages (> 1.5 mV), but all were within areas of abnormal innervation. This first feasibility study in humans confirms preclinical data and shows that the combination of neuronal imaging maps with more traditional anatomical voltage mapping could pave the way to more successful outcomes in VT ablation. Indeed, particularly in the setting of advanced SHD, VTs are often either not inducible or not hemodynamically tolerated in the procedural setting. Therefore, approaches to enhance our ability to identify arrhythmogenic substrate in patients are greatly needed.
Extracardiac neuronal structures also undergo a significant remodeling in cardiac diseases. In complete agreement with preclinical data [32, 33], the left stellate ganglia (LSG) of 31 patients with ischemic and non-ischemic CM suffering refractory Vas showed a significant neuronal enlargement combined with an increase in synaptic density [34]. More recently, inflammation, neurochemical remodeling, oxidative stress, and satellite glial cell activation were also described in the resected sympathetic ganglia of 16 patients with electrical storm and SHD undergoing cardiac sympathetic denervation (CSD) [35•]. Taken together, the preclinical and clinical data show that there is bilateral SG remodeling after spatially confined myocardial lesions, independent of the lesion site.
In SHDs, there are multiple triggers leading to increased neuronal activity and oxidative stress in the stellate ganglia that in turn may promote ganglionic inflammation. Overactivation of the CSAR combined with increased efferent sympathetic tone via a spinal reflex mechanism is thought to be the most important. Circulating neurohormonal signals such as angiotensin II [36, 37] or brainstem-mediated increases in efferent sympathetic outflow [38] may contribute as well.
Of note, mild but distinct inflammatory changes (CD3+ and CD8+ T cells and macrophages infiltrates) were reported in the LSG of 12 patients with channelopathies (long QT syndrome, LQTS, and catecholaminergic polymorphic VT, CPVT) and refractory VAs, together with degenerative changes of ganglion cells [39]. Neuronal number and neuronal size were not assessed in this study. Despite being very preliminary, these findings suggest that a T cell–mediated cytotoxicity toward ganglion cells may be involved in triggering and/or enhancing electrical instability also in structurally normal hearts with intrinsic arrhythmic susceptibility. Intriguingly, a subtle but clear mechanical dysfunction (both systolic and diastolic) in LQTS has been described since the 1990s [40] and subsequently confirmed using advanced echocardiographic techniques [41]. This feature is associated with both QT duration on EKG [42] and arrhythmic risk in LQTS [43] and may suggest a mechanism of functional CSAR overactivation in the absence of myocardial scar and/or left ventricular enlargement. In CPVT, as compared to LQTS, there is a consistently higher burden of premature ventricular complexes (PVCs) induced by catecholamine with a typically reproducible threshold. PVCs were recently shown to represent a strong drive for increased afferent signaling and neuronal remodeling in the ICNS [44].
Clinical Approaches to Neuromodulation in the Treatment of VAs
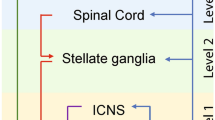
The strong association with the genesis of arrhythmias combined with the relatively accessible anatomical location has rendered several levels of the ANS valuable clinical targets for the management of patients with VAs. Figure 1 summarizes the main clinical approaches to autonomic modulation with antiarrhythmic purposes that will be described in the text.

Summary of the main interventions on the neuraxis to treat cardiac arrhythmias described in the text, divided by anatomical levels. ICNS, intrinsic cardiac nervous system
Thoracic Epidural Anesthesia and Spinal Cord Stimulation
At the level of the spinal cord, thoracic epidural anesthesia (TEA) and spinal cord stimulation (SCS) reduce spinal cord influences proximal to the SG either by injection of anesthetics or electrical stimulation, respectively.
TEA is performed by percutaneously injecting local anesthetics into the thoracic epidural space, thus pharmacologically blocking the T1-T5 spinal cord and roots and inhibiting both afferent and efferent sympathetic signaling to the heart. TEA causes prolongation of repolarization and refractory periods at the level of the myocardium [45, 46]. In a small study, TEA led to greater than 80% reduction in VAs in 6/8 patients [47]. Unfortunately, the effects of TEA are limited by the temporary duration of local anesthetics.
SCS has long been studied in the treatment of angina [48, 49], yet we are only recently recognizing its effects in reduction of VAs. SCS is achieved by inserting one or two leads with eight electrodes into the epidural space at the thoracic level. Prior canine and porcine studies have shown significant reduction in VAs even with brief (~ 1 h) periods of SCS [50, 51]. At present, data on SCS and VAs in humans are limited to case series and small clinical trials. In 2012, a case series of two patients who underwent SCS showed reduced VAs burden, from 128 and 90 episodes of VT over 2 months before, to 6 and 0 episodes over 2 months after SCS, respectively [52]. Two randomized control trials designed to evaluate effect of SCS on heart failure have had conflicting results. The SCS HEART study [53] showed a significant reduction in HF symptoms, NYHA class, and improvement of LVEF with SCS; however, the DEFEAT-HF study [54] did not. There was also no significant reduction in VAs based on limited data from ICD interrogations in the DEFEAT-HF study. The conflicting results could possibly be explained by the different anatomical locations (T1-T3 in SCS HEART versus T2-T4 in DEFEAT-HF) of the electrodes in the thoracic spinal cord, and/or by the different durations of SCS (continuously via implanted stimulator in DEFEAT-HF versus 12 h per day at 50 Hz for SCS HEART).
Stellate Ganglion Modulatory Therapy
Experimental data has consistently shown that stellate ganglion block/removal in animals strongly dampens the CSAR, providing protection against life-threatening VAs during acute myocardial ischemia [55], in the setting of a healed myocardial infarction [56, 57•] and in structurally normal hearts [58]. Not surprisingly, electrical stimulation of, as well as nerve growth factor (NGF) infusion into, the stellate ganglion (SG) is a powerful pro-arrhythmic trigger [59, 60].
In humans, the SG-targeted therapy tested so far includes cardiac sympathetic denervation (CSD), percutaneous blockade with local anesthetics (SGB), and radiofrequency ablation. Surgical removal of left thoracic sympathetic ganglia from T1 to T4 (cervicothoracic sympathectomy), also known as LCSD, was first performed more than one century ago by Dr. Jonnesco [61] for angina pectoris and cardiac arrhythmias. Subsequently, LCSD was also shown to be an effective antiarrhythmic strategy in LQTS [62] and CPVT [63••].
LCSD surgery was initially performed through a traditional, open approach, either via thoracotomy or, more frequently, via supraclavicular access [64]. More recently, a minimally invasive approach using video-assisted thoracic surgery (VATS) has been validated [65] and is now the most commonly used. This approach is considerably less invasive and leaves almost no visible scar in patients. Moreover, it enables a better visualization of the sympathetic chain, improving the capability of the surgeon to correctly identify the morphology of the thoracic gaglia [66] and the potential presence of anatomical variants such as Kunz’s nerve [67]. The only drawback of VATS is the higher incidence of transient neuropathic pain, rarely observed with the supraclavicular approach. Nevertheless, in a recent survey [68], patients with channelopathies treated with VATS LCSD showed a high degree of satisfaction with the procedure, even when it was performed for primary prevention.
More recently, CSD was implemented as an antiarrhythmic strategy in patients with SHD [69, 70] suffering from VAs refractory to drugs and catheter ablation. In this setting, bilateral CSD (BCSD) was shown to have a greater efficacy than LCSD in reducing not only appropriate ICD shocks [71] but also the combined endpoint of VA recurrence, mortality, and heart transplantation [72••]. In the largest study of CSD in SHD published to date, 121 patients underwent CSD, and the median number of ICD shocks per subject dropped from 10 in the year before to 0 in the year after CSD [72••]. This retrospective study consisted of patients with advanced cardiomyopathy (mean left ventricular ejection fraction 30%), and almost half of the participants had poor functional status (New York Heart Association functional class III or IV). Of note, despite bilateral CSD, 40% of these patients suffered recurrences of ICD shocks at 1 year, and the multivariate analysis showed that poor functional status and longer VT cycle lengths were the only predictors of both recurrent ICD shocks and the combined endpoint of sustained VT/ICD shock recurrence, death, and transplantation. Although the relationship between advanced heart failure and VAs is extremely complex, these results stress the importance of early consideration of CSD in patients with refractory VAs, particularly in those with good functional status, as already demonstrated for the referral to VT ablation procedures [73].
Due to its unique anatomical location, the SG can also be targeted in an emergency setting for the management of refractory VAs and electrical storms. Pharmacological SGB using anatomical landmarks has been safely performed since 1934 [74] for the treatment of sympathetic-related pain syndromes involving the upper extremities. Recent advances include an ultrasound-guided approach [75] as well as physical SGB using pulsed radiofrequency [76]. A systematic review [77] showed that percutaneous, transient, block of the SG through local anesthetic injection was associated with an acute reduction in VAs burden in 92% of subjects, with a decline from an average of 12 to 1 VA episodes per day. The majority (95%) of the 38 patients included in the study had an underlying cardiomyopathy (mean left ventricular ejection fraction 31%) and only 4 patients received a bilateral SGB. Therefore, in patients with SHDs, the acute protective effects of left SGB seem to be higher than the chronic effect of BCSD.
Although the mechanisms of VA recurrences after BCSD are complex, multifactorial, and possibly variable depending on the underlying myocardial substrate, it is becoming clear that in SHDs the entire neuraxis undergoes a progressive and dramatic remodeling, involving the afferent, sympathetic, and parasympathetic components at different levels spanning from the ICNS to the central nervous system [78, 79]. As such, the implementation of neuromodulatory interventions at earlier stages of SHDs as well as a combined approach acting at more levels of the neuraxis may be more beneficial in preventing arrhythmias, particularly in the middle and long term.
Stimulation of the Vagus Nerve and Its Auricular Branch
The most effective way of reproducibly increasing vagal activity is through direct electrical stimulation of the nerve at the cervical level, termed vagal nerve stimulation (VNS). In animal models, VNS significantly reduced VAs susceptibility after coronary artery occlusion and reperfusion [80] and in the setting of a healed myocardial infarction [81]. Moreover, VNS also modulated the cycle length, amplitude, and organization index of VAs [82]. Of note, preferential efferent, rather than afferent, fiber activation has been shown to be dependent on the shape and the orientation of the stimulating electrode as well as the frequencies, pulse widths, and currents used [83, 84]. Very recently, the antiarrhythmic effects of preferentially efferent VNS were evaluated in pigs in the setting of chronic MI and after BCSD [85•]. VT was inducible in all infarcted animals post-BCSD during isoproterenol infusion and VNS reduced inducibility by 67%. The study confirmed that the beneficial electrophysiological effects of VNS persist after BCSD.
Despite these promising findings, cervical VNS—though already approved in humans for the treatment of refractory epilepsy [86] and depression [87] and under evaluation for chronic heart failure [88••]—is invasive and not yet applicable in an acute clinical setting. A noninvasive alternative is now available, accessing the superficial collaterals of the vagus nerve’s auricular branch (ABVNS) from the ear. In canine models, chronic intermittent ABVNS at 2 h per day for 2 months reduced VAs inducibility, LSG neuronal activity, and sympathetic nerve sprouting in the border zone [89]. The effects of ABVNS on VAs in humans were recently reported in a randomized study [90•]. ABVNS was applied for 2 h in patients with ST elevation MI undergoing percutaneous reperfusion. The incidence of reperfusion-related VAs during the first 24 hours was significantly reduced, as well as infarct size and the inflammatory markers.
Interventions on ICNS
Due to the anatomical location and the clear pathophysiological role in the initiation and maintenance of atrial arrhythmias, the disruption of GPs has so far been proposed only for prevention of atrial fibrillation (AF) both in preclinical [91] and clinical studies [92], with conflicting results in paroxysmal/persistent AF [93•]. On the other hand, preliminary data suggest that the transient block of GPs through botulinum toxin injection into epicardial fat pads may provide long-term protection against post operatory AF [94]. Preclinical data also suggest that ablation of GPs may be pro-arrhythmic, leading to both atrial [95] and VAs [96]. From a clinical standpoint, a good example is provided by orthotropic cardiac transplantation. Despite small differences in surgical technique, the transplanted heart has a preserved ICNS from the donor, while the extracardiac neuronal connection of the recipient (afferent and efferent, both parasympathetic and sympathetic) is interrupted. The ICNS is capable of supporting the transplanted heart function on a beat-to-beat basis, while the risk of both VAs [97] and AF [98] is extremely low as compared with normal hearts. The conflicting nature of the data highlights the limitations to our overall knowledge about ICNS physiology.
Renal Denervation
Another area in need of greater study is how renal afferent signaling affects cardiac efferent sympathetic input from the CNS. While there have been many decades of studies on the effects of altering renal innervation on natriuresis and sympathetically driven hypertension [99••], less is known about the effects on cardiac arrhythmias. Renal artery sympathetic fibers run parallel to the artery ostially to distally in the adventitia. Denervation via catheter ablation along the renal artery (RAD) is thought to inhibit the afferent renal sympathetic pathway, which in turn would decrease efferent sympathetic influence on the heart [45]. RAD has shown promise in decreasing the burden of VA in a porcine model [99], as well as in small human studies. In 2015, an observational study of 10 patients with refractory VAs who underwent RAD showed that VA episodes and ICD shocks were both reduced from 28.5 and 8 before RAD to 0 and 0, respectively, after RAD [100]. A later retrospective study of 32 patients showed that the number of VA episodes, as well as ICD shocks and anti-tachycardia pacing episodes decreased significantly with catheter ablation of VT plus adjunctive RAD, compared with ablation alone [101]. There was, however, no mortality difference between the two arms.
Conclusions
The two divisions of the cardiac ANS, namely the sympathetic and the parasympathetic branches, play a primary role in ventricular arrhythmogenesis and have been extensively studied in the last decades. While the molecular effects of NE and Ach at the myocardial level have been largely elucidated, less is known about the anatomical and functional organization of the cardiac ANS and the mutual interaction between its two branches. Indeed, the neuraxis is proving to be an extremely complex and plastic entity, actively involved in the progression of the majority of heart diseases from their early stages. Hopefully in the near future, the ongoing dramatic progression in preclinical research tools, combined with larger clinical trials, will allow us to further validate and expand the application of autonomic modulation strategies for the treatment of heart rhythm disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Armour JA, Murphy DA, Yuan BX, Mac Donald S, Hopkins DA. Gross and microscopic anatomy of the human intrinsic cardiac nervous system. Anat Rec. 1997;247:289–98.
Vincentz JW, Rubart M, Firulli AB. Ontogeny of cardiac sympathetic innervation and its implications for cardiac disease. Pediatr Cardiol. 2012;33:923–8. https://doi.org/10.1007/s00246-012-0248-1.
Ardell JL, Randall WC, Cannon WJ, Schmacht DC, Tasdemiroglu E. Differential sympathetic regulation of automatic, conductile, and contractile tissue in dog heart. Am J Phys. 1988;1988(255):H1050–9. https://doi.org/10.1152/ajpheart.1988.255.5.H1050.
Ajijola OA, Vaseghi M, ZhouW YK, Benharash P, Hadaya J, et al. Functional differences between junctional and extrajunctional adrenergic receptor activation in mammalian ventricle. Am J Physiol Heart Circ Physiol. 2013;304:H579–88. https://doi.org/10.1152/ajpheart.00754.2012.
Andresen MC, Kuntz DL, Mendelowitz D. In: Armour JA, Ardell JL, editors. Central nervous system regulation of the heart. New York: Oxford University Press; 2004. p. 187–219.
Hopkins DA, Andrew AJ. Ganglionic distribution of afferent neurons innervating the canine heart and cardiopulmonary nerves. J Auton Nerv Syst. 1989;26:213–22.
Armour JA. Potential clinical relevance of the “little brain” on the mammalian heart. Exp Physiol. 2008;93:165–76. https://doi.org/10.1113/expphysiol.2007.041178.
Pauza DH, Skripka V, Pauziene N, Stropus R. Morphology, distribution, and variability of the epicardiac neural ganglionated subplexuses in the human heart. Anat Rec. 2000;259:353–82.
Charpentier F, Drouin E, Gauthier C, LeMarec H. Early after/depolarizations and triggered activity: mechanisms and autonomic regulation. Fundam Clin Pharmacol. 1993;7:39–49.
Zipes DP, Barber MJ, Takahashi N, Gilmour RF Jr. Influence of the autonomic nervous system on the genesis of cardiac arrhythmias. Pacing Clin Electrophysiol. 1983;6:1210–20.
Ajijola OA, Lux RL, Khahera A, Kwon O, Aliotta E, Ennis DB, et al. Sympathetic modulation of electrical activation in normal and infarcted myocardium: implications for arrhythmogenesis. Am J Physiol Heart Circ Physiol. 2017;312:H608–21. https://doi.org/10.1152/ajpheart.00575.2016.
Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res. 2014;114:1004–21. https://doi.org/10.1161/CIRCRESAHA.113.302549.
Yagishita D, Chui RW, Yamakawa K, Rajendran PS, Ajijola OA, Nakamura K, et al. Sympathetic nerve stimulation, not circulating norepinephrine, modulates T-peak to T-end interval by increasing global dispersion of repolarization. Circ Arrhythm Electrophysiol. 2015;8:174–85. https://doi.org/10.1161/CIRCEP.114.002195.
Vanhoutee PM, Verbeuren TJ. Inhibition by acetylcholine of the norepinephrine release evoked by potassium in canine saphenous veins. Circ Res. 1976;39:263–9.
Shanks J, Herring N. Peripheral cardiac sympathetic hyperactivity in cardiovascular disease: role of neuropeptides. Am J Physiol Regul Integr Comp Physiol 2013;305:R1411–20. https://doi.org/10.1152/ajpregu.00118.2013
Calvillo L, Vanoli E, Andreoli E, Besana A, Omodeo E, Gnecchi, et al. Vagal stimulation, through its nicotinic action, limits infarct size and the inflammatory response to myocardial ischemia and reperfusion. J Cardiovasc Pharmacol. 2011;58:500–7. https://doi.org/10.1097/FJC.0b013e31822b7204.
Floras JS. Sympathetic nervous system activation in human heart failure. Clinical implications of an updated model. J Am Coll Cardiol. 2009;54:375–85. https://doi.org/10.1016/j.jacc.2009.03.061.
Schwartz PJ, Pagani M, Lombardi F, Malliani A, Brown AM. A cardio-cardiac sympatho-vagal reflex in the cat. Circ Res. 1973;32:215–20.
Cerati D, Schwartz PJ. Single cardiac vagal fibers activity, acute myocardial ischemia, and risk for sudden death. Circ Res. 1991;69:1389–401.
Zahner MR, Li DP, Chen SR, Pan HL. Cardiac vanilloid receptor 1-expressing afferent nerves and their role in the cardiogenic sympathetic reflex in rats. J Physiol. 2003;2003(551):515–23. https://doi.org/10.1113/jphysiol.2003.048207.
Pan HL, Chen SR. Sensing tissue ischemia: another new function for capsaicin receptors? Circulation. 2004;110:1826–31. https://doi.org/10.1161/01.CIR.0000142618.20278.7A.
Uchida Y, Murao S. Bradykinin-induced excitation of afferent cardiac sympathetic nerve fibers. Jpn Heart J. 1974;15:84–91. https://doi.org/10.1536/ihj.15.84.
Schultz HD, Ustinova EE. Capsaicin receptors mediate free radical-induced activation of cardiac afferent endings. Cardiovasc Res. 1998;38:348–55. https://doi.org/10.1016/S0008-6363(98)00031-5.
Wang W, Schultz HD, Ma R. Cardiac sympathetic afferent sensitivity is enhanced in heart failure. Am J Physiol Heart Circ Physiol. 1999;277:H812–7.
Wang HJ, Wang W, Cornish KG, Rozanski GJ, Zucker IH. Cardiac sympathetic afferent denervation attenuates cardiac remodeling and improves cardiovascular dysfunction in rats with heart failure. Hypertension. 2014;64:745–55. https://doi.org/10.1161/HYPERTENSIONAHA.114.03699.
Cha YM, Redfield MM, Shah S, Shen WK, Fishbein MC, Chen PS. Effects of omapatrilat on cardiac nerve sprouting and structural remodeling in experimental congestive heart failure. Heart Rhythm. 2005;2:984–90. https://doi.org/10.1016/j.hrthm.2005.05.016.
Inoue H, Zipes DP. Results of sympathetic denervation in the canine heart: supersensitivity that may be arrhythmogenic. Circulation. 1987;75:877–87.
Inoue H, Zipes DP. Time course of denervation of efferent sympathetic and vagal nerves after occlusion of the coronary artery in the canine heart. Circ Res. 1988;62:1111–20.
Vaseghi M, Lux RL, Mahajan A, Shivkumar K. Sympathetic stimulation increases dispersion of repolarization in humans with myocardial infarc- tion. Am J Physiol Heart Circ Physiol. 2012;302:H1838–46. https://doi.org/10.1152/ajpheart.01106.2011.
Verma A, Marrouche NF, Schweikert RA, Saliba W, Wazni O, Cummings J, et al. Relationship between successful ablation sites and the scar border zone defined by substrate mapping for ventricular tachycardia post-myocardial infarction. J Cardiovasc Electrophysiol. 2005;16:465–71. https://doi.org/10.1046/j.1540-8167.2005.40443.x.
• Klein T, Abdulghani M, Smith M, Huang R, Asoglu R, Remo BF, et al. Three-Dimensional 123I-Meta-Iodobenzylguanidine Cardiac Innervation Maps to Assess Substrate and Successful Ablation Sites for Ventricular Tachycardia: Feasibility Study for a Novel Paradigm of Innervation Imaging. Circ Arrhythm Electrophysiol. 2015;8:583–91. https://doi.org/10.1161/CIRCEP.114.002105. First in humans study who demonstrated the possibility of using 123I-Meta-Iodobenzylguanidine to build three-dimensional innervation maps of the heart to guide VT ablation procedures.
Han S, Kobayashi K, Joung B, Piccirillo G, Maruyama M, Vinters HV, et al. Electroanatomic remodeling of the left stellate ganglion after myocardial infarction. J Am Coll Cardiol. 2012;59:954–61. https://doi.org/10.1016/j.jacc.2011.11.030.
Ajijola OA, Yagishita D, Reddy NK, Yamakawa K, Vaseghi M, Downs AM, et al. Remodeling of stellate ganglion neurons after spatially targeted myocardial infarction: neuropeptide and morphologic changes. Heart Rhythm. 2015;12:1027–35. https://doi.org/10.1016/j.hrthm.2015.01.045.
Ajijola OA, Wisco JJ, Lambert HW, Mahajan A, Stark E, Fishbein MC, et al. Extracardiac neural remodeling in humans with cardiomyopathy. Circ Arrhythm Electrophysiol. 2012;5:1010–116. https://doi.org/10.1161/CIRCEP.112.972836.
• Ajijola OA, Hoover DB, Simerly TM, Brown TC, Yanagawa J, Biniwale RM, et al. Inflammation, oxidative stress, and glial cell activation characterize stellate ganglia from humans with electrical storm. JCI Insight. 2017;2:e94715. https://doi.org/10.1172/jci.insight.94715. Detailed in vitro evaluation of the stellate ganglia from humans with structural heart disease and electrical storm showing neuronal remodeling.
Xiao L, Haack KK, Zucker IH. Angiotensin II regulates ACE and ACE2 in neurons through p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling. Am J Phys Cell Phys. 2013;304:C1073–9. https://doi.org/10.1152/ajpcell.00364.2012.
Zucker IH, Gao L. The regulation of sympathetic nerve activity by angiotensin II involves reactive oxygen species and MAPK. Circ Res. 2005;97:737–9. https://doi.org/10.1161/01.RES.0000188261.94569.1f.
Lambert GW, Kaye DM, Lefkovits J, Jennings GL, Turner AG, Cox HS, et al. Increased central nervous system monoamine neurotransmitter turnover and its association with sympathetic nervous activity in treated heart failure patients. Circulation. 1995;92:1813–8.
Rizzo S, Basso C, Troost D, Aronica E, Frigo AC, Driessen AH, et al. T-cell-mediated inflammatory activity in the stellate ganglia of patients with ion-channel disease and severe ventricular arrhythmias. Circ Arrhythm Electrophysiol. 2014;7:224–9. https://doi.org/10.1161/CIRCEP.113.001184.
Nador F, Beria G, De Ferrari GM, Stramba-Badiale M, Locati EH, Lotto A, et al. Unsuspected echocardiographic abnormality in the long QT syndrome: diagnostic, prognostic, and pathogenetic implications. Circulation. 1991;84:1530–42.
Haugaa KH, Amlie JP, Berge KE, Leren TP, Smiseth OA, Edvardsen T. Transmural differences in myocardial contraction in long-QT syndrome: mechanical consequences of ion channel dysfunction. Circulation. 2010;122:1355–63. https://doi.org/10.1161/CIRCULATIONAHA.110.960377.
Leren IS, Hasselberg NE, Saberniak J, Håland TF, Kongsgård E, Smiseth OA, et al. Cardiac mechanical alterations and genotype specific differences in subjects with long QT syndrome. J Am Coll Cardiol Img. 2015;8:501–10. https://doi.org/10.1016/j.jcmg.2014.12.023.
Haugaa KH, Edvardsen T, Leren TP, Gran JM, Smiseth OA, Amlie JP. Left ventricular mechanical dispersion by tissue Doppler imaging: a novel approach for identifying high-risk individuals with long QT syndrome. Eur Heart J. 2009;30:330–7. https://doi.org/10.1093/eurheartj/ehn466.
Hamon D, Rajendran PS, Chui RW, Ajijola OA, Irie T, Talebi R, et al. Premature ventricular contraction coupling interval variability destabilizes cardiac neuronal and electrophysiological control: insights from simultaneous cardioneural mapping. Circ Arrhythm Electrophysiol. 2017;10. https://doi.org/10.1161/CIRCEP.116.004937.
Tung R, Shivkumar K. Neuraxial modulation for treatment of VT storm. J Biomed Res. 2015;29:56–60. https://doi.org/10.7555/JBR.29.20140161.
Howard-Quijano K, Takamiya T, Dale EA, Kipke J, Kubo Y, Grogan T, et al. Spinal cord stimulation reduces ventricular arrhythmias during acute ischemia by attenuation of regional myocardial excitability. Am J Physiol Heart Circ Physiol. 2017;313:H421–31. https://doi.org/10.1152/ajpheart.00129.2017.
Bourke T, Vaseghi M, Michowitz Y, Sankhla V, Shah M, Swapna N, et al. Neuraxial modulation for refractory ventricular arrhythmias: value of thoracic epidural anesthesia and surgical left cardiac sympathetic denervation. Circulation. 2010;121:2255–62. https://doi.org/10.1161/CIRCULATIONAHA.109.929703.
Mannheimer C, Camici P, Chester MR, Collins A, DeJongste M, Eliasson T, et al. The problem of chronic refractory angina; report from the ESC Joint Study Group on the Treatment of Refractory Angina. Eur Heart J. 2002;23:355–70.
Zhang TC, Janik JJ, Grill WM. Mechanisms and models of spinal cord stimulation for the treatment of neuropathic pain. Brain Res. 2014;1569:19–31. https://doi.org/10.1016/j.brainres.2014.04.039.
Wang S, Zhou X, Huang B, Wang Z, Liao K, Saren G, et al. Spinal cord stimulation protects against ventricular arrhythmias by suppressing left stellate ganglion neural activity in an acute myocardial infarction canine model. Heart Rhythm. 2015;12:1628–35. https://doi.org/10.1016/j.hrthm.2015.03.023.
Odenstedt J, Linderoth B, Bergfeldt L, Ekre O, Grip L, Mannheimer C, et al. Spinal cord stimulation effects on myocardial ischemia, infarct size, ventricular arrhythmia, and noninvasive electrophysiology in a porcine ischemia–reperfusion model. Heart Rhythm. 2011;8:892–8. https://doi.org/10.1016/j.hrthm.2011.01.029.
Grimaldi R, de Luca A, Kornet L, Castagno D, Gaita F. Can spinal cord stimulation reduce ventricular arrhythmias? Heart Rhythm. 2012;9:1884–7. https://doi.org/10.1016/j.hrthm.2012.08.007.
Tse HF, Turner S, Sanders P, Okuyama Y, Fujiu K, Cheung CW, et al. Thoracic Spinal Cord Stimulation for Heart Failure as a Restorative Treatment (SCS HEART study): first-in-man experience. Heart Rhythm. 2015;12:588–95. https://doi.org/10.1111/j.1540-8167.2011.02230.x.
Zipes DP, Neuzil P, Theres H, Caraway D, Mann DL, Mannheimer C, et al. Determining the feasibility of spinal cord neuromodulation for the treatment of chronic systolic heart failure: the DEFEAT-HF study. JACC Heart Fail. 2016;4:129–36. https://doi.org/10.1016/j.jchf.2015.10.006.
Schwartz PJ, Stone HL, Brown AM. Effects of unilateral stellate ganglion blockade on the arrhythmias associated with coronary occlusion. Am. Heart J. 1976;92:589–99.
Schwartz PJ, Billman GE, Stone HL. Autonomic mechanisms in ventricular fibrillation induced by myocardial ischemia during exercise in dogs with healed myocardial infarction: an experimental preparation for sudden cardiac death. Circulation. 1984;69:790–800.
• Irie T, Yamakawa K, Hamon D, Nakamura K, Shivkumar K, Vaseghi M. Cardiac sympathetic innervation via middle cervical and stellate ganglia and antiarrhythmic mechanism of bilateral stellectomy. Am J Physiol Heart Circ Physiol. 2017;312:H392–405. https://doi.org/10.1152/ajpheart.00644.2016. First demonstration in a big animal model of the rule of middle cervical ganglia in regulating electrical properties of the heart and cardiovascular reflexes.
Schwartz PJ, Snebold NG, Brown AM. Effects of unilateral cardiac sympathetic denervation on the ventricular fibrillation threshold. Am J Cardiol. 1976;37:1034–40.
Schwartz PJ, Malliani A. Electrical alternation of the T wave: clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long QT syndrome. Am. Heart J. 1975;89:45–50.
Cao JM, Chen LS, KenKnight BH, Ohara T, Lee MH, Tsai J, et al. Nerve sprouting and sudden cardiac death. Circ Res. 2000;86:816–21.
Schwartz PJ, De Ferrari GM, Pugliese L. Cardiac sympathetic denervation 100 years later: Jonnesco would have never believed it. Int J Cardiol. 2017;237:25–8. https://doi.org/10.1016/j.ijcard.2017.03.020.
Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long QT syndrome. Circulation. 2004;109:1826–33. https://doi.org/10.1161/01.CIR.0000125523.14403.1E.
•• De Ferrari GM, Dusi V, Spazzolini C, Bos JM, Abrams DJ, Berul CI, et al. Clinical management of catecholaminergic polymorphic ventricular tachycardia: the role of left cardiac sympathetic denervation. Circulation. 2015;131:2185–93. https://doi.org/10.1161/CIRCULATIONAHA.115.015731. The largest human study showing the efficacy of LCSD in VAs in CVPT.
Odero A, Bozzani A, De Ferrari GM, Schwartz PJ. Left cardiac sympathetic denervation for the prevention of life-threatening arrhythmias: the surgical supraclavicular approach to cervicothoracic sympathectomy. Heart Rhythm. 2010;7:1161–5. https://doi.org/10.1016/j.hrthm.2010.03.046.
Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009;6:752–9. https://doi.org/10.1016/j.hrthm.2009.03.024.
Kwon OJ, Pendekanti S, Fox JN, Yanagawa J, Fishbein MC, Shivkumar K, et al. Morphological spectra of adult human stellate ganglia: implications for thoracic sympathetic denervation. Anat Rec. 2018. https://doi.org/10.1002/ar.23797.
Zaidi ZF, Ashraf A. The nerve of Kunz: incidence, location and variations. J Appl Sci Res. 2010;6:659–64.
Waddell-Smith KE, Ertresvaag KN, Li J, Chaudhuri K, Crawford JR, Hamill JK, et al. Physical and psychological consequences of left cardiac sympathetic denervation in long-QT syndrome and catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2015;8:1151–8. https://doi.org/10.1161/CIRCEP.115.003159.
Coleman MA, Bos MJ, Johnson JN, Owen HJ, Deschamps C, Moir C, et al. Videoscopic left cardiac sympathetic denervation for patients with recurrent ventricular fibrillation/malignant ventricular arrhythmia syndromes besides congenital long-QT syndrome. Circ Arrhythm Electrophysiol. 2012;5:782–8. https://doi.org/10.1161/CIRCEP.112.971754.
Ajijola OA, Lellouche N, Bourke T, Tung R, Ahn S, Mahajan A, et al. Bilateral cardiac sympathetic denervation for the management of electrical storm. J Am Coll Cardiol. 2012;59:91–2. https://doi.org/10.1016/j.jacc.2011.09.043.
Vaseghi M, Gima J, Kanaan C, Ajijola OA, Marmureanu A, Mahajan A, et al. Cardiac sympathetic denervation in patients with refractory ven- tricular arrhythmias or electrical storm: intermediate and long-term follow-up. Heart Rhythm. 2014;11:360–6. https://doi.org/10.1016/j.hrthm.2013.11.028.
•• Vaseghi M, Barwad P, Malavassi Corrales FJ, Tandri H, Mathuria N, et al. Cardiac sympathetic denervation for refractory ventricular arrhythmias. J Am Coll Cardiol. 2017;69:3070–80. https://doi.org/10.1016/j.jacc.2017.04.035. The largest and most recent study showing that CSD decreased VAs in patients with structural heart disease.
Romero J, Di Biase L, Diaz JC, Quispe R, Du X, Briceno D, et al. Early versus late referral for catheter ablation of ventricular tachycardia in patients with structural heart disease. JACC. 2018;3:374–82. https://doi.org/10.1016/j.jacep.2017.12.008.
Leriche R, Fontaine R. L’Anesthe ́ sie isole ́ e du ganglion e ́ toile ́ . Sa technique, ses indications, ses re ́ sultats. Presse Med. 1934;42:849–50.
Gofeld M, Bhatia A, Abbas S, Ganapathy S, Johnson M. Development and validation of a new technique for ultrasound-guided stellate ganglion block. Reg Anesth Pain Med. 2009;34:475–9. https://doi.org/10.1097/AAP.0b013e3181b494de.
Hayase J, Vampola S, Ahadian F, Narayan SM, Krummen DE. Comparative efficacy of stellate ganglion block with bupivacaine vs pulsed radiofrequency in a patient with refractory ventricular arrhythmias. J Clin Anesth. 2016;31:162–5. https://doi.org/10.1016/j.jclinane.2016.01.026.
Meng L, Tseng CH, Shivkumar K, Ajijola O. Efficacy of stellate ganglion blockade in managing electrical storm: a systematic review. JACC Clin Electrophysiol. 2017;9:942–9. https://doi.org/10.1016/j.jacep.2017.06.006.
Kumar R, Woo MA, Birrer BV, Macey PM, Fonarow GC, Hamilton MA, et al. Mammillary bodies and fornix fibers are injured in heart failure. Neurobiol Dis. 2009;33:236–42. https://doi.org/10.1016/j.nbd.2008.10.004.
Woo MA, Palomares JA, Macey PM, Fonarow GC, Harper RM, Kumar R. Global and regional brain mean diffusivity changes in patients with heart failure. J Neurosci Res. 2015;93:678–85. https://doi.org/10.1002/jnr.23525.
Zuanetti G, De Ferrari GM, Priori SG, Schwartz PJ. Protective effect of vagal stimulation on reperfusion arrhythmias in cats. Circ Res. 1987;61:429–35.
Vanoli E, De Ferrari GM, Stramba-Badiale M, Hull SS Jr, Foreman RD, Schwartz PJ. Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ Res. 1991;68:1471–81.
Nazeri A, Elayda MA, Dragnev L, Frank CM, Qu J, Afonso VX, et al. Heterogeneity of left ventricular signal characteristics in response to acute vagal stimulation during ventricular fibrillation in dogs. Tex Heart Inst J. 2011;38:621–6.
Ardell JL, Rajendran PS, Nier HA, KenKnight BH, Armour JA. Central-peripheral neural network interactions evoked by vagus nerve stimulation: functional consequences on control of cardiac function. Am J Physiol Heart Circ Physiol. 2015;309:H1740–52. https://doi.org/10.1152/ajpheart.00557.2015.
Yoo PB, Lubock NB, Hincapie JG, Ruble SB, Hamann JJ, Grill WM. High-resolution measurement of electrically-evoked vagus nerve activity in the anesthetized dog. J Neural Eng. 2013;10:026003. https://doi.org/10.1088/1741-2560/10/2/026003.
• Yamaguchi N, Yamakawa K, Rajendran PS, Takamiya T, Vaseghi M. Antiarrhythmic effects of vagal nerve stimulation after cardiac sympathetic denervation in the setting of chronic myocardial infarction. Heart Rhythm. 2018;15:1214–22. https://doi.org/10.1016/j.hrthm.2018.03.012. First animal study showing the beneficial antiarrhythmic effects of VNS after CSD in the setting of chronic myocardial infarction.
Uthman BM, Reichl AM, Dean JC, Eisenschenk S, Gilmore R, Reid S, et al. Effectiveness of vagus nerve stimulation in epilepsy patients: a 12-year observation. Neurology. 2004;63:1124–6.
Shuchman M. Approving the vagus-nerve stimulator for depression. N Engl J Med. 2007;356:1604–7. https://doi.org/10.1056/NEJMp078035.
•• Shivkumar K, Ajijola OA, Anand I, Armour JA, Chen PS, Esler M, et al. Clinical neurocardiology defining the value of neuroscience-based cardiovascular therapeutics. J Physiol. 2016;594:3911–54. https://doi.org/10.1113/JP271870. First position paper by international experts in the field defining the rationale and the clinical impact of neuroscience-based cardiovascular therapeutics.
Yu L, Wang S, Zhou X, Wang Z, Huang B, Liao K, et al. Chronic intermittent low-level stimulation of tragus reduces cardiac autonomic remodeling and ventricular arrhythmia inducibility in a post-infarction canine model. JACC Clin Electrophysiol. 2016;2:330–9. https://doi.org/10.1016/j.jacep.2015.11.006.
• Yu L, Huang B, Po SS, Tan T, Wang M, Zhou L, et al. Low-Level tragus stimulation for the treatment of ischemia and reperfusion injury in patients with ST-segment elevation myocardial infarction: a proof-of-concept study. JACC Cardiovasc Interv. 2017;10:1511–20. https://doi.org/10.1016/j.jcin.2017.04.036. First in humans, randomized study showing the beneficial effect of low-level tragus stimulation for the treatment of ischemia and reperfusion injury.
Shen MJ, Shinohara T, Park HW, Frick K, Ice DS, Choi EK, et al. Continuous low-level vagus nerve stimulation reduces stellate ganglion nerve activity and paroxysmal atrial tachyarrhythmias in ambulatory canines. Circulation. 2011;123:2204–12. https://doi.org/10.1161/CIRCULATIONAHA.111.018028.
Katritsis DG, Pokushalov E, Romanov A, Giazitzoglou E, Siontis GC, Po SS, et al. Autonomic denervation added to pulmonary vein isolation for paroxysmal atrial fibrillation: a randomized clinical trial. JAmColl Cardiol. 2013;62:2318–25. https://doi.org/10.1016/j.jacc.2013.06.053.
• Driessen AH, Berger WR, Krul SP, Vanden Berg NW, Neefs J, Piersma FR, et al. Ganglion plexus ablation in advanced atrial fibrillation: the AFACT study. J Am Coll Cardiol. 2016;68:1155–65. https://doi.org/10.1016/j.jacc.2016.06.036 .The largest, randomized clinical trial of ganglion plexus ablation in advanced atrial fibrillation, with negative finding.
Pokushalov E, Kozlov B, Romanov A, Strelnikov A, Bayramova S, Sergeevichev D, et al. Long-term suppression of atrial fibrillation by botulinum toxin injection into epicardial fat pad in patients undergoing cardiac surgery: one-year follow-up of a randomized pilot study. Circ Arrhythm Electrophysiol. 2015;8:1334–41. https://doi.org/10.1161/CIRCEP.115.003199.
Lo LW, Scherlag BJ, Chang HY, Lin YJ, Chen SA, Po SS. Paradoxical long-term proarrhythmic effects after ablating the “head station” ganglionated plexi of the vagal innervation to the heart. Heart Rhythm. 2013;10:751–7. https://doi.org/10.1016/j.hrthm.2013.01.030.
He B, Lu Z, He W, Wu L, Cui B, Hu X, et al. Effects of ganglionated plexi ablation on ventricular electrophysiological properties in normal hearts and after acute myocardial ischemia. Int J Cardiol. 2013;168:86–93. https://doi.org/10.1016/j.ijcard.2012.09.067.
Vaseghi M, Lellouche N, Ritter H, Fonarow GC, Patel JK, Moriguchi J, et al. Mode and mechanisms of death after orthotopic heart transplantation. Heart Rhythm. 2009;6:503–9. https://doi.org/10.1016/j.hrthm.2009.01.005.
Vaseghi M, Boyle NG, Kedia R, Patel JK, Cesario DA, Wiener I, et al. Supraventricular tachycardia after orthotopic cardiac transplantation. J Am Coll Cardiol. 2008;51:2241–9. https://doi.org/10.1016/j.jacc.2008.02.065.
Jackson N, Gizurarson S, Azam MA, King B, Ramadeen A, Zamiri N, et al. Effects of renal artery denervation on ventricular arrhythmias in a postinfarct model. Circ Cardiovasc Interv. 2017;10:e004172. https://doi.org/10.1161/CIRCINTERVENTIONS.116.004172.
Armaganijan LV, Staico R, Moreira DA, Lopes RD, Medeiros PT, Habib R, et al. 6-month outcomes in patients with implantable cardioverter-defibrillators undergoing renal sympathetic denervation for the treatment of refractory ventricular arrhythmias. JACC Cardiovasc Interv. 2015;8:984–90. https://doi.org/10.1016/j.jcin.2015.03.012.
Evranos B, Canpolat U, Kocyigit D, Coteli C, Yorgun H, Aytemir K, et al. Role of adjuvant renal sympathetic denervation in the treatment of ventricular arrhythmias. Am J Cardiol. 2016;118:1207–10. https://doi.org/10.1016/j.amjcard.2016.07.036.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Veronica Dusi, Ching Zhu, and Olujimi A. Ajijola declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This is a review article so we have referenced studies done in humans and animals, but these studies were not done as part of this article, and they are just referenced here. For those studies, all procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. And all applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Invasive Electrophysiology and Pacing
Rights and permissions
About this article

Cite this article
Dusi, V., Zhu, C. & Ajijola, O.A. Neuromodulation Approaches for Cardiac Arrhythmias: Recent Advances. Curr Cardiol Rep 21, 32 (2019). https://doi.org/10.1007/s11886-019-1120-1
Published:
DOI: https://doi.org/10.1007/s11886-019-1120-1