
Abstract
In the USA, 69 % of adults are either overweight or obese and 35 % are obese. Obesity is associated with an increased incidence of various cardiovascular disorders. Obesity is a risk marker for cardiovascular disease, in that it is associated with a much higher prevalence of comorbidities such as diabetes, hypertension, and metabolic syndrome, which then increase the risk for cardiovascular disease. However, in addition, obesity may also be an independent risk factor for the development of cardiovascular disease. Furthermore, although obesity has been shown to be an independent risk factor for several cardiovascular diseases, it is often associated with improved survival once the diagnosis of the cardiovascular disease has been made, leading to the term “obesity paradox.” Several pathways linking obesity and cardiovascular disease have been described. In this review, we attempt to summarize the complex relationship between obesity and cardiovascular disorders, in particular coronary atherosclerosis, heart failure, and atrial fibrillation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the past two decades, obesity has become a global epidemic affecting both pediatric and adult populations. In the USA, 69 % of adults are either overweight or obese and 35 % are obese [1]. Obesity has been associated with a decreased life expectancy as well as with increased morbidity [2]. The relationship between cardiovascular disease (CV) and obesity has been widely studied, but a number of questions still remain. For instance, obesity has been linked to development of cardiovascular diseases including atherosclerosis and symptomatic coronary artery disease (CAD), heart failure (HF), and atrial fibrillation (Fig. 1). Obesity does increase the risk of CV disease secondarily through its influence on the development and severity of comorbidities such as hypertension, dyslipidemia, and glucose intolerance or diabetes [3]. However, the increase in various CV diseases may also occur in the absence of other comorbidities and may be due to structural and functional changes of the myocardium through excess adipose tissue deposition or through other mechanisms related to obesity [4]. Furthermore, obesity has been known to be an independent risk factor for the development of CV diseases such as HF but has also been shown to be associated with improved survival once the diagnosis of HF is established (the so-called “obesity paradox”) [5, 6•, 7]. This obesity paradox has also been observed in other CV disease states [8].

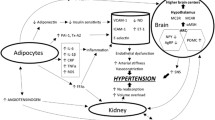
Increased risk of cardiovascular diseases with increase in body mass index (BMI) and body weight. CAD = coronary artery disease; HF = heart failure; SBP = systolic blood pressure; DBP = diastolic blood pressure
In this review, we attempt to emphasize the association of obesity with CV disease, focusing on whether obesity is an independent risk factor for the development of CV disease states such as CAD, HF, and atrial fibrillation. The fact that obesity is a risk marker, in that it is associated with a much higher prevalence of comorbidities such as diabetes, hypertension, and metabolic syndrome, which themselves are risk factors for the development of CV disease, is well established and will not be discussed in any detail.
Measures of Obesity
Several measures other than just body weight are used to measure obesity, including body mass index (BMI), waist circumference, and waist-to-hip ratio. Body weight indexed to height to measure the BMI is a commonly used index of obesity and overweight. An adult is considered normal weight if the BMI is 18.5 to <25 kg/m2, overweight if the BMI is 25.0 to 29.9 kg/m2, and obese if the BMI is ≥30.0 kg/m2 [9]. Waist circumference has been shown to be a better index of abdominal obesity than waist-to-hip ratio. It also correlates better with BMI than waist-to-hip ratio [10]. A recent study showed overall age-adjusted obesity prevalence to be 35.7 %. There were, however, race-specific age-adjusted differences noted; specifically, 36.2 % non-Hispanic white men, 38.8 % non-Hispanic black men, 32.2 % non-Hispanic white women, and 58.5 % non-Hispanic black women met the BMI criteria for obesity [1].
Obesity and Atherosclerotic Disease
The understanding of the pathophysiology of atherosclerosis and obesity has dramatically changed over the past few decades. Historically, obesity and atherosclerosis were thought to be simply lipid storage disorders involving triglycerides in adipose tissue and cholesteryl ester in atheromata. However, now they are both also viewed as chronic inflammatory disease states, with activation of both innate and adaptive immunity [11•, 12].
Atherosclerotic disease and obesity share several common pathophysiological features. First, lipids contribute to both atherosclerosis and obesity; oxidized low-density lipoprotein (LDL) and free fatty acids can trigger inflammation and initiate disease. Inflammation mediates all stages of atherogenesis—from early lesion development to atheroma complication—and is associated with obesity, insulin resistance, and type 2 diabetes mellitus. Inflammation may form the major link between obesity and atherosclerosis: Adipokines released by adipose tissue induce insulin resistance, endothelial dysfunction, hypercoagulability, and systemic inflammation, all of which can promote atherosclerosis. The accumulation of heterogeneous macrophage populations, T cell activation, cell death, and the effects of numerous cytokines and chemokines characterize both atherosclerosis and obesity. Inflammatory biomarkers, such as high-sensitivity C-reactive protein, IL-6, and IL-18, can predict cardiovascular events, may be used to guide therapy, and reflect the pathophysiological links between obesity and its associated metabolic disorders [9]. When evaluated in a clinical trial, a multidisciplinary program, which aimed to reduce body weight in obese women through lifestyle changes, was associated with a reduction in markers of vascular inflammation (CRP, IL-6, and IL-18) and in insulin resistance along with an increase in levels of adiponectin, an adipocytokine with anti-inflammatory and insulin-sensitizing properties [13].
It is important to understand the link between inflammation and atherosclerosis and how obesity accelerates this process. Obese individuals have a higher propensity toward inflammation compared to non-obese individuals. Patients with visceral obesity have been found to have higher levels of proinflammatory adipokines including TNF-alpha, IL-6, MCP-1, resistin, and leptin [14]. The higher level of inflammation has been correlated with observations from several large-scale prospective studies that demonstrated elevated levels of CRP in obese patients [15]. Elevated CRP levels independently predict a higher risk of future myocardial infarction, peripheral arterial disease, and the risk of developing type II diabetes mellitus [16–18]. Furthermore, CRP levels correlate with increased visceral adiposity independent of overall BMI [19]. This has led to the use of CRP levels in patients with intermediate risk of coronary artery disease to improve risk stratification for major adverse cardiovascular events (MACEs) [20].
As the evidence of atherosclerosis being a chronic inflammatory state increased, so did the therapeutic approach to its treatment. HMG-CoA reductase inhibitors or statins were initially developed to decrease LDL cholesterol, but many studies have shown that they also provide an anti-inflammatory effect including the reduction of CRP levels [21, 22]. Using this background, the Justification for the Use of Statins in Primary Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial demonstrated that the number of cardiovascular events was significantly decreased in healthy individuals with an LDL cholesterol less than 130 mg/dl but with CRP levels of 2 mg/l or higher, through the use of 20 mg rosuvastatin daily compared with placebo [23]. Similarly, salicylates such as aspirin (which of course also have additional anti-platelet action) have been known to decrease inflammation and have been used to treat many different conditions ranging from rheumatoid arthritis to primary prevention of myocardial infarction. Studies, however, have found that the use of non-acetylated salicylate derivative, salsalates, which do not increase bleeding times, decreases inflammation and improves glycemic control in obese individuals [24–26]. Anti-inflammatory drugs could be beneficial in improving CV outcomes in obese atherosclerotic patients, but further long-term studies are necessary.
The relationship between obesity and CAD can be assessed in two settings. The first is obesity’s role in the pathogenesis of coronary atherosclerosis and stable ischemic heart disease and, second, its implications in acute coronary syndromes (ACSs) and coronary revascularization. It is known that obesity is an independent risk factor for CAD [3], as well as for ACS when CAD is present [27].
The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study allowed a better understanding of the relationship between obesity and CAD [28]. In this study, the investigators conducted a post-mortem study on the arteries of young individuals who had died from accidental causes or suicides. They concluded that the onset of CAD occurs decades before the actual clinical manifestations. They also determined that higher BMI was associated with more complex coronary lesions, as shown by an increased number of fatty streaks, raised lesions, and high-grade complex lesions, when compared to normal weight individuals. Another study showed that the complexity of coronary lesions was directly related to panniculus thickness in all patients with BMI >30 kg/m2 [28]. In addition, obesity likely needs to be long-standing and present for at least two decades to become an independent risk factor for clinically significant coronary artery disease, as suggested by several long-term cohort studies [29–31]. One study showed that a 10-kg increase in body weight was associated with 12 % increased risk of CAD and 3 mmHg higher systolic and 2.3 mmHg higher diastolic blood pressure [32].
As detailed earlier, the pathogenesis of CAD in obese individuals is mediated via adipose tissue, which serves as an active paracrine and endocrine organ, and produces a wide array of adipokines that are directly involved in atherosclerosis [33]. These adipokines interact and influence several other metabolic and immunologic cascades, such as vascular reactivity (e.g., leptin, TNF-α, and adiponectin), inflammation (e.g., monocyte chemotactic protein 1 and IL-8) and coagulation (e.g., plasminogen activator inhibitor). These pathways often overlap and activate more bioactive factors, following a positive feedback course [34–37]. In comparison to normal weight individuals, circulating platelets in obese counterparts show increased activation as well as diminished sensitivity to anti-platelet agents such as aspirin [38, 39]. Despite all this knowledge, the comprehensive cellular and molecular pathways linking obesity to development of CAD and triggering of ACS are not completely understood. Fat distribution appears to play a key role. Abdominal obesity is associated with worse outcomes in patients with known CVD, as demonstrated by data from the Trandolapril Cardiac Evaluation (TRACE) registry [40]. Moreover, this relationship remained significant after adjustment for comorbidities associated with obesity, such as diabetes and hypertension. In the INTERHEART study, waist-to-hip ratio, waist circumference, and waist-to-height ratio were all stronger predictors of myocardial infarction (MI) compared to BMI alone [41]. This indicates that the “abdominal” component of obesity has the most detrimental effects pertaining to CAD in obese individuals.
Obesity and Acute Cardiovascular Syndromes
In a large study including more than 100,000 patients who presented with non-ST elevation myocardial infarction (NSTEMI), obesity was found to be the strongest factor linked to NSTEMI events in younger patients, followed by tobacco use. The higher the BMI, the lower the mean age at which the patients presented with NSTEMI [42]. This inverse correlation with age also holds true for ST elevation myocardial infarction (STEMI) patients [43]. Based on the available data, obesity is considered an independent risk factor for STEMI in younger patients [44, 45]. Moreover, obesity is also linked to development of other acute vascular events; for each 1-unit increase in BMI, there is a 4 % increased risk of ischemic stroke and 6 % increased risk for hemorrhagic stroke [32, 46].
Obesity and Revascularization for CAD
In addition to obesity being a risk factor for the development of ACS, obesity may also increase the risk of adverse events after treatment for these conditions. In a follow-up study of patients who received bare metal stents, obesity was found to be an independent predictor of increased in-stent restenosis, 1-year target revascularization, and MACE [47]. The Limus Eluted from a Durable Versus Erodable Stent Coating (LEADERS) trial showed that obesity was an independent risk factor for stent thrombosis [48]. However, a large European cohort of over 7000 patients did not show any association of BMI and stent thrombosis or adverse cardiac events [49]. The neutral effect of obesity in patients with drug eluting stents has been shown in other studies as well [50, 51]. Based on this data, whether drug-eluting stents should be given even greater preference in all obese patients remains debatable. There is also controversial data regarding obesity and resistance to thienopyridines, and the clinical relevance of studies linking obesity with clopidogrel resistance is yet to be confirmed [52].
Obesity Paradox in Patients with CAD
Surprisingly, despite the observations linking obesity with increased incidence of acute myocardial infarction (NSTEMI or STEMI), several large studies have shown obese patients to have better survival when examining mortality after MI (obesity paradox). Data from large ACS trials such as the Superior Yield of the New Strategy of Enoxaparin, Revascularization, and Glycoprotein IIb/IIIa Inhibitors (SYNERGY) [53] and Metabolic Efficiency with Ranolazine for Less Ischaemia in NSTE-ACS (MERLIN)-TIMI 36 [54] have shown an independent inverse correlation between BMI and overall mortality. A recent meta-analysis of 10 post–percutaneous coronary intervention (PCI) and 12 post–coronary artery bypass grafting (CABG) studies further substantiates the obesity paradox after coronary revascularization. In both post-PCI and post-CABG subgroup of patients, the 30-day all-cause mortality was lower in obese patients compared to their normal weight counterparts [55]. The protective effect of obesity, however, seems to decline when severe or morbid obesity is present (J-shaped relationship), and the lowest mortality may be seen in overweight patients [56].
Obesity and Heart Failure
Over the last few decades, there have been significant advancements in available medical and interventional therapies in cardiology. Despite the use of several mortality-reducing therapies, the number of cases of incident HF and of acute decompensated HF hospitalizations has increased [57]. During the same time, incidence of obesity has also been on the rise. As noted previously, 35 % of the US population is obese, and approximately 69 % are either overweight or obese [1]. There exists a well-established link between obesity and HF; hence, the rising prevalence of both obesity and HF may make this association an important target for prevention. One of the first published reports linking obesity and HF in 1956 was a case report of a morbidly obese female who had “heart failure” from the compressive effects of the physical mass of the adipose tissue underneath her chest wall [58]. We now know that obesity has several complex mechanisms by which it may lead to HF. The term “obesity cardiomyopathy” was first used in 1992, when an analysis of 519 patients, using right heart catheterization and endomyocardial biopsy, showed that individuals with higher BMI were more likely to have dilated cardiomyopathy than their lean counterparts [59].
Epidemiological Association of Obesity with Heart Failure
Kenchaiah et al. reported the first large epidemiological study showing obesity to be an independent risk factor for development of HF, analyzing 5881 individuals from the Framingham Heart Study. It was concluded that for each increment of 1 kg/m2 in BMI, there was an increase in the risk of HF of 7 % for women and 5 % for men [60]. As compared with subjects with a normal BMI, obese individuals had a doubling of the risk of HF. For women, the hazard ratio was 2.17 (95 % confidence interval (CI), 1.51 to 2.97); for men, the hazard ratio was 1.90 (95 % CI, 1.30 to 2.79). The risk for development of HF was independent of age, alcohol, and cigarette use and comorbidities including but not limited to diabetes, hypertension, and history of myocardial infarction. Similarly, an analysis from the Physician’s Health Study of 21,094 men without known coronary artery disease showed that compared to normal weight, overweight and obesity were independently associated with an increase of HF [61]. Loehr et al. examined the Atherosclerosis Risk in Communities cohort of over 14,000 individuals and showed obesity to be an independent risk factor for development of HF, after adjusting the covariates [62]. Similarly, another large study of over 59,000 individuals from Finland showed a graded association between BMI and HF risk, with adjusted hazard ratios of HF for overweight and obese patients as compared to normal weight of 1.25 (95 % CI = 1.12–1.39) and 1.99 (95 % CI = 1.74–2.27) in males, and 1.33 (95 % CI = 1.16–1.51) and 2.06 (95 % CI = 1.80–2.37) in females [63]. In addition to BMI, other anthropometric indices of obesity including waist circumference, waist-to-hip ratio, and waist-to-height ratio have also been independently associated with incident HF in large, population-based studies; however, indices such as waist circumference and waist-to-hip ratio have not been shown to perform better than BMI as predictors of HF [62, 64].
Protective Effect of Physical Activity
It should be noted that physical activity appears to influence the relationship between obesity and HF. In the Physicians Health Study, in addition to increasing BMI, lower level of physical activity was also associated with an increased risk of HF, with the highest relative risk of HF seen in obese men who were also physically inactive compared to men who were lean and physically active [61]. Similarly, in the Finnish cohort discussed above, participants demonstrated the protective effect of physical activity at all levels of obesity, reducing risk of HF in fully adjusted models by 21–32 % [63].
Mechanisms of Obesity-Associated Heart Failure
There are several possible theories to explain the effect of higher BMI leading to development of HF (Fig. 2). Obesity can produce a range of hemodynamic changes that can predispose to changes in cardiac morphology and ventricular function, including left ventricular (LV) dilation, eccentric or concentric LV hypertrophy, LV systolic and diastolic dysfunction, and RV dysfunction [65, 66]. Obesity has also been described as a state of hemodynamic overload associated with increase in cardiac output and blood pressure [67]. Over a long duration, this could lead to left ventricular hypertrophy, depressed systolic function, and impaired relaxation [68, 69]. While these changes occur in all classes of obesity, they are most pronounced in the severely obese and could potentially lead to HF in such individuals. Another mechanism involves a direct effect of excess body fat on the myocardium causing cardiac adaptation and remodeling, which has often been termed obesity cardiomyopathy [70, 71]. Although the concept of a cardiomyopathy related to obesity has previously been described, severe left ventricular systolic dysfunction occurs uncommonly due to obesity alone and its presence should trigger an investigation for other contributory factors, before attributing it to obesity alone. On the other hand, HF with preserved left ventricular ejection fraction (HFpEF) is seen much more frequently in obese patients [72, 73].

Interplay of various mechanisms in obese individuals that may lead to the development of heart failure. SVR = systemic vascular resistance; CO = cardiac output; CAD = coronary artery disease; MI = myocardial infarction; RVH = right ventricular hypertrophy; LV = left ventricular
Although the relationship between obesity and incident HF may be related to hemodynamic and anatomic cardiac changes related to excess body mass, evidence suggests that the relationship is also mediated by obesity-related metabolic, inflammatory, and neurohormonal changes. Obesity and insulin resistance are highly correlated, which may in part potentiate the link between obesity and HF. In the Uppsala study, insulin sensitivity but not anthropometric indices of obesity was independently predictive of HF risk when both were evaluated together in a fully adjusted model [74]. Abnormalities in the adipokine pathway, as well as other inflammatory cytokines seen in obese individuals, may contribute to the pathophysiology of HF [70, 75]. The adipokine resistin, expressed by adipocytes and associated with insulin resistance and inflammation, was associated with the risk of developing HF independent of coronary heart disease and other risk factors in a Framingham offspring analysis [76]. Of note, leptin has been studied in great detail. Produced by adipocytes, its levels have been found to directly correlate with BMI and waist circumference. Higher levels have been associated with an increased risk for incident HF in patients without pre-existing coronary artery disease, although this association was absent in patients with pre-existing CAD [75]. The role of inflammation is also supported by an analysis from the Multi-Ethnic Study of Atherosclerosis (MESA). Although the risk of HF was 83 % higher in obese compared to that in non-obese subjects after adjustment for traditional risk factors, the relationship between obesity and incident HF was no longer significant after adjustment for the inflammatory biomarkers, IL-6 or C-reactive protein [77]. Finally, as is well known, obesity is associated with the other comorbid risk factors that contribute to incident HF, such as DM, HTN, and CAD [68].
Obesity Paradox in HF
Several studies have shown that despite being an independent risk factor for the development of HF, obesity is associated with lower mortality in patients with established HF, the “obesity paradox in HF” [5, 6•, 78]. Several hypotheses have been suggested to explain this apparent paradox [6•, 79]. Severe HF due to its catabolic effect may be associated with weight loss, known as “cardiac cachexia,” and was initially thought to be the explanation for better survival in overweight or obese HF patients. Although the obesity paradox in established HF has been demonstrated in several studies, a more recent study found that that even overweight and obesity prior to incident HF (i.e., pre-morbid or pre-HF higher body mass index) are associated with improved survival compared to normal weight up to 10 years after development of HF, suggesting that weight loss due to advanced HF may not completely explain the protective effect of higher body mass index (BMI) in HF patients [80•].
Several neuro-humoral pathways have also been postulated to explain the obesity paradox in HF. In obese patients, high levels of serum lipoproteins may neutralize bacterial toxins or circulating cytokines, such as TNF-α [81]. Obese patients also have decreased adiponectin levels and low catecholamine response, both of which may be associated with improved HF survival [79]. Levels of circulating stem cells are also increased in obese patients, which may lead to improved outcomes [82]. Furthermore, obese patients are likely to be diagnosed early with HF due to the symptoms produced by excess body weight, such as dyspnea and edema. This may lead to an apparently improved life span, i.e., lead time bias [80•]. Obese patients have multiple comorbidities, such as diabetes mellitus and hypertension, and hence represent a high-risk screening population. Moreover, given higher blood pressures in the overweight and obese population groups, greater up-titration of disease modifying therapies could be possible. All these hypotheses could explain the reasons behind the observed obesity paradox in HF. Interestingly, a recent study found that the obesity paradox was not seen in overweight male patients after adjustment for confounding variables but remained in overweight female patients [83]. The protective effect in female patients could be partially explained by recent research which suggests that female hearts have greater myocardial fatty acid uptake and lesser myocardial glucose utilization in advanced HF [83]. Future studies are needed to further understand the pathways involved.
Currently, the most recent HF guidelines by the American College of Cardiology/American Heart Association do not specifically recommend weight reduction in obese patients with HF [84]. Large, prospective, randomized control trials with long follow-up would be able to definitively answer the effectiveness of intentional weight loss interventions in obese HF patients. Nevertheless, based on the data available, obese HF patients appear to benefit from exercise interventions.
Atrial Fibrillation
Atrial fibrillation (AF) is one of the most common arrhythmias worldwide and is more commonly observed in obese individuals [85]. A report from the Framingham study showed that after adjustment for CV disease risk factors, and the occurrence of interim myocardial infarction or HF, every 1-unit increment in BMI was independently associated with a 4 % increase in risk of AF in both men and women [85] (Fig. 1). When examined by BMI categories of obesity, the investigators found that compared to normal weight individuals, obese individuals had a 50 % increased risk of AF. The Danish Diet, Cancer, and Health Cohort Study also demonstrated a higher risk of AF associated with increased anthropometric measurements such as height, weight, BMI, hip circumference, and waist circumference, as well as with increased bioimpedance-derived measures of body fat mass, body fat percentage, and lean body mass [86].
Mechanisms for Atrial Fibrillation with Obesity
The Framingham study demonstrated that the excess risk of AF associated with obesity appeared to be mediated by left atrial dilatation. [87]. The investigators observed a graded increase in left atrial size as BMI category increased from normal to overweight to obese, consistent with previous observations of obesity as a major risk factor for the development of left atrial enlargement [88, 89]. After adjusting for left atrial diameter, obesity was no longer associated with an increased risk for the development of AF, whereas left atrial diameter remained strongly associated with risk of AF. Left atrial dilation/remodeling which is an established mechanistically important factor in the pathogenesis of AF is likely multifactorial in obesity and occurs as a result of increased central blood volume, and secondary to elevated left ventricular diastolic pressures both due to obesity-driven load with increase in left ventricular mass and HF, as well as secondary to obesity-associated hypertension, diabetes, and obstructive sleep apnea (Fig. 3) [90•].

Interplay of various mechanisms in obese individuals that may lead to the development of atrial fibrillation. CAD = coronary artery disease; DM = diabetes mellitus; OSA = obstructive sleep apnea; HTN = hypertension
Interestingly, genetic studies have suggested a link between non-valvular AF and obesity. Specifically, polymorphisms of TaqIB cholesteryl transfer protein gene and 1444 C/T of CRP gene have been shown to be associated with an increased susceptibility in obese males [91]. Additionally, a G protein-coupled potassium channel known as GIRK4 (G protein-coupled inward rectifier K channel), which is found prevalently in the heart, has been shown to be abnormally expressed in obese individuals with AF [92].
Several recent studies have described the relationship between thickness of epicardial adipose tissue (EAT) and AF burden [91]. A significant correlation exists between EAT and BMI, waist circumference, or visceral adipose tissue [93]. The Framingham study reported that pericardial fat volume predicted AF, independently of other measures of adiposity, including BMI [94]. Furthermore, studies indicate that pericardial fat is associated with the increased prevalence and severity of AF, independent of traditional risk factors, including left atrial dilation [95], suggesting that EAT volume may have prognostic significance besides traditional measures of obesity [96, 97]. It has been speculated that the direct impact of obesity on atrial substrate may be mediated via EAT [96]. As detailed before, adipose tissue produces a variety of bioactive molecules, such as inflammatory mediators and adipocytokines that have been shown to mediate the effect of visceral fat on other tissues. Among others, EAT also secretes activin A and matrix metalloproteinases (MMPs), which may mediate the fibrotic effect of EAT on the atrial wall [98]. Inflammatory markers such as CRP, TNF-α, IL-2, IL-6, IL-8, and monocyte chemoattractant protein (MCP)-1 have been associated with AF [99] and are produced and secreted by EAT, specifically in the setting of obesity, diabetes, and ischemic cardiomyopathy. There is also increasing evidence that implicates inflammation in AF development and perpetuation. In addition, EAT accumulation is associated with fatty infiltration from the epicardial layer, which advances deep into the myocardium, contributing to myocardial functional disorganization and the formation of local arrhythmogenic substrate [100]. EAT may also affect the atrial cellular components by providing a source for precursor cells that can differentiate into myofibroblasts and thus contribute to atrial remodeling, a substrate for AF [101].
Furthermore, patients with lower BMI and a single episode of new-onset AF have a lower risk of progressing to permanent AF compared to obese individuals [98]. Interestingly, a recent trial in Australia comparing the Long-Term Effect of Goal Directed Weight Management in an Atrial fibrillation Cohort (LEGACY) found that long-term weight loss is associated with a significant decrease in the incidence of recurrent AF [102]. In LEGACY, the individuals with ≥10 % weight loss resulted in a sixfold greater probability of AF-free survival [102]. The risk of developing hypertension, diabetes, metabolic syndrome, coronary artery disease, and sleep apnea is expectedly significantly higher in obese individuals. Therefore, the risk of AF is also increased in obese patients through the various mechanisms that contribute to the development of AF in patients with these comorbidities [90•].
Conclusion and Future Directions
In summary, we have discussed the evidence that supports obesity as both an independent risk factor and a risk marker for the development of asymptomatic and symptomatic coronary artery disease, heart failure, and atrial fibrillation. Future studies exploring the role of weight loss as a positive disease modifier in these conditions are urgently needed.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Flegal K, Carroll M, Kit B, Ogden C. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307:491–7.
Fontaine K, Redden D, Wang C, Westfall A, Allison D. Years of life lost due to obesity. JAMA. 2003;289:187–93.
Poirier P, Giles T, Bray G, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Circulation. 2006;113:898–918.
Poirier P, Martin J, Marceau P, Biron S, Marceau S. Impact of bariatric surgery on cardiac structure, function and clinical manifestations in morbid obesity. Expert Rev Cardiovasc Ther. 2004;2:193–201.
Horwich TB, Fonarow GC, Hamilton M, MacLellan W, Woo M, Tillisch J. The relationship between obesity and mortality in patients with heart failure. J Am Coll Cardiol. 2001;38:789–95.
Lavie CJ, Alpert MA, Arena R, Mehra MR, Milani RV, Ventura HO. Impact of obesity and the obesity paradox on prevalence and prognosis in heart failure. JACC Heart Fail. 2013;1(2):93–102. Lavie et al. review the data on obesity and cardiopulmonary fitness in heart failure patients. The article also includes informative data on obesity and its impact on treatments such as heart transplantation and LV assist devices.
Shah R, Gayat E, Januzzi Jr JL, et al. Mebazaa A Body mass index and mortality in acutely decompensated heart failure across the world: a global obesity paradox. J Am Coll Cardiol. 2014;63:778–85.
Akin I, Nienaber C. “Obesity paradox” in coronary artery disease. World J Cardiol. 2015;7(10):603–8.
Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults: The Evidence Report: National Institutes of Health. Obes Res. 1998;2:51S–209S.
Kurpad SS, Tandon H, Srinivasan K. Waist circumference correlates better with body mass index than waist-to-hip ratio in Asian Indians. Natl Med J India. 2003;16(4):189–92.
Rocha V, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. 2009;6:399–409. Rocha et al. review in detail the interplay between obesity and inflammation leading to the development of atherosclerosis.
Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340(2):115–26.
Esposito K, Pontillo A, Di Palo C, et al. Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA. 2003;289:1799–804.
Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132:2169–80.
Ridker P. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49:2129–38.
Everett B, Kurth T, Buring J, Ridker P. The relative strength of C-reactive protein and lipid levels as determinants of ischemic stroke compared with coronary heart disease in women. J Am Coll Cardiol. 2006;48:2235–42.
Ridker P, Stampfer M, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA. 2001;285(19):2481–5.
Pradhan A, Manson J, Rifai N, Buring J, Ridker P. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–34.
Visser M, Bouter L, McQuillan G, Wener M, Harris T. Elevated C-reactive protein levels in overweight and obese adults. JAMA. 1999;282:2131–5.
Pearson T, Mensah G, Alexander R, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511.
Ridker P, Rifai N, Pfeffer M, et al. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events (CAre) investigators. Circulation. 1998;98:839–44.
Ridker P, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344:1959–65.
Ridker P, Danielson E, Fonseca F, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207.
Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity and diet-induced insulin resistance with salicylates or targeted disruption of ikkbeta. Science. 2001;293:1673–7.
Fleischman A, Shoelson S, Bernier R, Goldfine A. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care. 2008;31:289–94.
Koska J, Ortega E, Bunt J, et al. The effect of salsalate on insulin action and glucose tolerance in obese non-diabetic patients: results of a randomised double-blind placebo-controlled study. Diabetologia. 2009;52:385–93.
Wolk R, Berger P, Lennon J, Brilakis E, Somers V. Body mass index: a risk factor for unstable angina and myocardial infarction in patients with angiographically confirmed coronary artery disease. Circulation. 2003;108:2206–11.
McGill H, McMahan C, Herderick E, et al. Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation. 2002;105:2712–8.
Rabkin S, Mathewson F, Hsu P. Relation of body weight to development of ischemic heart disease in a cohort of young North American men after a 26 year observation period: the Manitoba Study. Am J Cardiol. 1977;39:452–8.
Manson J, Colditz G, Stampfer M, et al. A prospective study of obesity and risk of coronary heart disease in women. N Engl J Med. 1990;322:882–9.
Wilson P, D’Agostino R, Sullivan L, Parise H, Kannel W. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Arch Intern Med. 2002;16:1867–72.
Din-Dzietham R, Liu Y, Bielo M, Shamsa F. High blood pressure trends in children and adolescents in national surveys, 1963 to 2002. Circulation. 2007;116(13):1488–96.
Lau D, Dhillon B, Yan H, Szmitko P, Verma S. Adipokines: molecular links between obesity and atheroslcerosis. Am J Physiol Heart Circ Physiol. 2005;288:2031–41.
Juge-Aubry C, Henrichot E, Meier C. Adipose tissue: a regulator of inflammation. Best Pract Res Clin Endocrinol Metab. 2005;19:547–66.
Knudson J, Dincer U, Zhang C, et al. Leptin receptors are expressed in coronary arteries and hyperleptinemia causes significant coronary endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2005;289:48–56.
Trayhurn P, Wood I. Signaling role of adipose tissue: adipokines and inflammation in obesity. Biochem Soc Trans. 2005;33:1078–81.
Payne G, Kohr M, Tune J. Epicardial perivascular adipose tissue as a therapeutic target in obesity-related coronary artery disease. Br J Pharmacol. 2012;165(3):659–69.
Anfossi G, Russo I, Trovati M. Platelet dysfunction in central obesity. Nutr Metab Cardiovasc Dis. 2009;19:440–9.
Salama M, Morad A, Saleh M, Sabri N, Zaki M, ElSafady L. Resistance to low-dose aspirin therapy among patients with acute coronary syndrome in relation to associated risk factors. J Clin Pharm Ther. 2012;37:630–6.
Kragelund C, Hassager C, Hildebrandt P, Torp-Pedersen C, Kober L. Impact of obesity on long-term prognosis following acute myocardial infarction. Int J Cardiol. 2005;98:123–31.
Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case–control study. Lancet. 2004;364:937–52.
Madala M, Franklin B, Chen A, et al. Obesity and age of first non-ST-segment elevation myocardial infarction. J Am Coll Cardiol. 2008;52:979–85.
Das S, Alexander K, Chen A, et al. Impact of body weight and extreme obesity on the presentation, treatment, and in-hospital outcomes of 50,149 patients with ST-Segment elevation myocardial infarction results from the NCDR (National Cardiovascular Data). J Am Coll Cardiol. 2011;58(25):2642–50.
Bajaj S, Shamoon F, Gupta N, et al. Acute ST-segment elevation myocardial infarction in young adults: who is at risk? Coron Artery Dis. 2011;22:238–44.
Jamil G, Jamil M, Alkhazraji H, et al. Risk factor assessment of young patients with acute myocardial infarction. Am J Cardiovasc Dis. 2013;3:170–4.
Lavie C, Milani R, Ventura H. Obesity and cardiovascular disease: risk factor, paradox and impact of weight loss. J Am Coll Cardiol. 2009;53(21):1925–32.
Nikolsky E, Kosinski E, Mishkel GJ, et al. Impact of obesity on revascularization and restenosis rates after bare-metal and drug-eluting stent implantation (from the TAXUS-IV trial). Am J Cardiol. 2005;95:709–15.
Sarno G, Garg S, Onuma Y, et al. The impact of body mass index on the one year outcomes of patients treated by percutaneous coronary intervention with biolimus- and sirolimus-eluting stents (from the LEADERS Trial). Am J Cardiol. 2010;105:475–9.
Sarno G, Raber L, Onuma Y, et al. Impact of body mass index on the five-year outcome of patients having percutaneous coronary interventions with drug-eluting stents. Am J Cardiol. 2011;108:195–201.
Akin I, Tolg R, Hochadel M, et al. No evidence of “obesity paradox” after treatment with drug-eluting stents in a routine clinical practice: results from the prospective multicenter German DES.DE (German Drug-Eluting Stent) Registry. JACC Cardivasc Interv. 2012;5(2):162–9.
Wang Z, Zhou Y, Zhao Y, et al. Effect of obesity on repeat revascularization in patients undergoing percutaneous coronary intervention with drug-eluting stents. Obesity (Silver Spring). 2012;20:141–6.
Feher G, Koltai K, Alkonyi B, et al. Clopidogrel resistance: role of body mass and concomitant medications. Int J Cardiol. 2007;120:188–92.
Mahaffey K, Tonev S, Spinler S, et al. Obesity in patients with non-ST-segment elevation acute coronary syndromes: results from the SYNERGY trial. Int J Cardiol. 2010;139:123–33.
Kadakia M, Fox C, Scirica B, Murphy S, Bonaca M, Morrow D. Central obesity and cardiovascular outcomes in patients with acute coronary syndrome: observations from the MERLIN-TIMI 36 trial. Heart. 2011;97:1782–7.
Oreopoulos A, Padwal R, Norris C, Mullen J, Pretorius V, Kalantar-Zadeh K. Effect of obesity on short- and long-term mortality postcoronary revascularization: a meta-analysis. Obesity (Silver Spring). 2008;16:442–50.
Li Y, Lin G, Lin C, Wang J, Han C. Relation of body mass index to mortality among patients with percutaneous coronary intervention longer than 5 years follow-up: a meta-analysis. Int J Cardiol. 2013;168:4315–8.
Bui A, Horwich T, Fonarow G. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8(1):30–41.
Counihan T. Heart failure due to extreme obesity. Br Heart J. 1956;18(3):425–6.
Kasper EK, Hruban RH, Baughman KL. Cardiomyopathy of obesity: a clinicopathologic evaluation of 43 obese patients with heart failure. Am J Cardiol. 1992;70(9):921–4.
Kenchaiah S, Evans J, Levy D, et al. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305–13.
Kenchaiah S, Sesso H, Gaziano J. Body mass index and vigorous physical activity and the risk of heart failure among men. Circulation. 2009;119(1):44–52.
Loehr L, Rosamond W, Poole C, et al. Association of multiple anthropometrics of overweight and obesity with incident heart failure: the Atherosclerosis Risk in Communities study. Circ Heart Fail. 2009;2(1):18–24.
Hu G, Jousilahti P, Antikainen R, Katzmarzyk PT, Tuomilehto J. Joint effects of physical activity, body mass index, waist circumference, and waist-to-hip ratio on the risk of heart failure. Circulation. 2010;121(2):237–44.
Hu G, Jousilahti P, Antikainen R, Katzmarzyk P, Tuomilehto J. Joint effects of physical activity, body mass index, waist circumference, and waist-to-hip ratio on the risk of heart failure. Circulation. 2010;121:237–44.
Pascual M, Pascual D, Soria F, et al. Effects of isolated obesity on systolic and diastolic left ventricular function. Heart. 2003;89(10):1152–6.
Alpert M, Alexander J. Obesity and ventricular function in man: systolic function. In: Alpert MA, Alexander JK, editors. The heart and lung in obesity. Armonk, New York: Futura Publishing Company; 1998. p. 77–94.
Alexander J, Dennis E, Smith W, Amad K, Duncan W, Austin R. Blood volume, cardiac output, and distribution of systemic blood flow in extreme obesity. Cardiovasc Res Cent Bull. 1962;1:39–44.
Alpert M, Lambert C, Panayiotou H, et al. Relation of duration of morbid obesity to left ventricular mass, systolic function, and diastolic filling, and effect of weight loss. Am J Cardiol. 1995;76:1194–7.
Nakajima T, Fujioka S, Tokunaga K, Hirobe K, Matsuzawa Y, Tarui S. Noninvasive study of left ventricular performance in obese patients: influence of duration of obesity. Circulation. 1985;71:481–6.
McGavock J, Victor R, Unger R, Szczepaniak L. Adiposity of the heart, revisited. Ann Intern Med. 2006;144:517–24.
Poirier P, Giles T, Bray G, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol. 2006;26:968–76.
Kapoor J, Heidenreich P. Obesity and survival in patients with heart failure and preserved systolic function: a U-shaped relationship. Am Heart J. 2010;159:75–80.
Ather S, Chan W, Bozkurt B, et al. Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. J Am Coll Cardiol. 2012;59(11):998–1005.
Ingelsson E, Sundstrom J, Arnlöv J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA. 2005;294(3):334–41.
Wannamethee S, Shaper A, Whincup P, Lennon L, Sattar N. Obesity and risk of incident heart failure in older men with and without pre-existing coronary heart disease: does leptin have a role? J Am Coll Cardiol. 2011;58:1870–7.
Frankel D, Vasan R, D’Agostino R, et al. Resistin, adiponectin, and risk of heart failure the Framingham offspring study. J Am Coll Cardiol. 2009;53(9):754–62.
Bahrami H, Bluemke D, Kronmal R, et al. Novel metabolic risk factors for incident heart failure and their relationship with obesity: the MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;51(18):1775–83.
Bozkurt B, Deswal A. Obesity as a prognostic factor in chronic symptomatic heart failure. Am Heart J. 2005;150(6):1233–9.
Kistorp C, Faber J, Galatius S, et al. Plasma adiponectin, body mass index, and mortality in patients with chronic heart failure. Circulation. 2005;112:1756–62.
Khalid U, Ather S, Bavishi C, et al. Pre-morbid body mass index and mortality after incident heart failure: the ARIC Study. J Am Coll Cardiol. 2014;64(25):2743–9. This is the first study that describes the obesity paradox in heart failure, based on pre-heart failure (i.e. premorbid) BMI. The authors found that obesity and underweight compared to normal weight prior to the development of heart failure was associated with improved survival after the development of heart failure.
Lavie C, Ventura H. Weighing in on obesity and the obesity paradox in heart failure. J Card Fail. 2011;17:381–3.
Blogowski W, Serwin K, Budkowska M, et al. Clinical analysis of systemic and adipose tissue levels of selected hormones/adipokines and stromal-derived factor-1. J Biol Regul Homeost Agents. 2012;26:607–15.
Vest A, Wu Y, Hachamovitch R, Young J, Cho L. The heart failure overweight/obesity survival paradox: the missing sex link. J Am Coll Cardiol HF. 2015;3:917–26.
Yancy C, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:147–239.
Wang T, Parise H, Levy D, et al. Obesity and the risk of new-onset atrial fibrillation. JAMA. 2004;292:2471–7.
Frost L, Benjamin E, Fenger-Grøn M, Pedersen A. Tjønneland A, Overvad K. Body fat, body fat distribution, lean body mass, and atrial fibrillation and flutter. A Danish cohort study. Obesity (Silver Spring). 2014;22:1546–52.
Wanahita N, Messerli F, Bangalore S, Gami A, Somers V, Steinberg J. Atrial fibrillation and obesity: results of a meta-analysis. Am Heart J. 2008;155:310–5.
Iacobellis G, Ribaudo M, Leto G, et al. Influence of excess fat on cardiac morphology and function: study in uncomplicated obesity. Obes Res. 2002;10:767–73.
Gottdiener J, Reda D, Williams D, Materson B. Left atrial size in hypertensive men: influence of obesity, race and age. J Am Coll Cardiol. 1997;29:651–8.
Goudis C, Korantzopoulos P, Ntalas I, Kallergis E, Ketikoglou D. Obesity and atrial fibrillation: a comprehensive review of the pathophysiological mechanisms and links. J Cardiol. 2015;66(5):361–9. Goudis et al. review the most recent clinical and experimental studies linking obesity to atrial fibrillation.
Yang W, Xu L, Zhang H. Association of CETP and CRP gene polymorphisms with non-valvular atrial fibrillation in Chinese Han population. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2008;25:225–229.
Kang Y, Hu Y, Li N. Advances in research on G protein-coupled inward rectifier K(+) channel gene. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2012;34:426–430.
Rabkin S. The relationship between epicardial fat and indices of obesity and the metabolic syndrome: a systematic review and meta-analysis. Metab Syndr Relat Disord. 2014;12:31–42.
Thanassoulis G, Massaro J, O’Donnell CJ, et al. Pericardial fat is associated with prevalent atrial fibrillation: the Framingham Heart Study. Circ Arrhythm E. 2010;3(4):345–50.
Al Chekakie M, Welles C, Metoyer R, et al. Pericardial fat is independently associated with human atrial fibrillation. J Am Coll Cardiol. 2010;56:784–8.
Wong C, Abed H, Molaee P, et al. Pericardial fat is associated with atrial fibrillation severity and ablation outcome. J Am Coll Cardiol. 2011;57(17):1745–51.
Shin S, Yong H, Lim H, et al. Total and interatrial epicardial adipose tissues are independently associated with left atrial remodeling in patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2011;22(6):647–55.
Hatem S, Sanders P. Epicardial adipose tissue and atrial fibrillation. Cardiovasc Res. 2014;102:205–13.
Guo Y, Lip G, Apostolakis S. Inflammation in atrial fibrillation. J Am Coll Cardiol. 2012;60:2263–70.
Mahajan R, Brooks A, Shipp N. Epicardial fat infiltration of atrial musculature creates the substrate for atrial fibrillation in obesity. Heart Rhythm. 2012;9:124.
Zuk P, Zhu M, Ashjian P, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13:4279–95.
Pathak R, Middeldorp M, Meredith M, Mehta A, MActSt, Mahajan R, Wong C, Twomey D, Elliott A, Kalman J, Abhayaratna W, Lau D, Sanders P. Long-Term Effect of Goal-Directed Weight Management in an Atrial Fibrillation Cohort. J Am Coll Cardiol. 2015;65:2159–2169
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Taher Mandviwala and Umair Khalid declare that they have no conflict of interest.
Anita Deswal declares research support from Novartis as site-PI of multicenter clinical trial, and grant support from the NIH as site-PI for clinical trials.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Coronary Heart Disease
Rights and permissions
About this article

Cite this article
Mandviwala, T., Khalid, U. & Deswal, A. Obesity and Cardiovascular Disease: a Risk Factor or a Risk Marker?. Curr Atheroscler Rep 18, 21 (2016). https://doi.org/10.1007/s11883-016-0575-4
Published:
DOI: https://doi.org/10.1007/s11883-016-0575-4