
Abstract
Purpose of Review
Cardiac sarcoidosis (CS) is an important cause of non-ischemic cardiomyopathy and has specific diagnostic and therapeutic considerations. With advances in imaging techniques and treatment approaches, the approach to monitoring disease progression and management of CS continues to evolve. The purpose of this review is to highlight advances in CS diagnosis and treatment and present a center’s multidisciplinary approach to CS care.
Recent Findings
In this review, we highlight advances in granuloma biology along with contemporary diagnostic approaches. Moreover, we expand on current targets of immunosuppression focused on granuloma biology and concurrent advances in the cardiovascular care of CS in light of recent guideline recommendations.
Summary
Here, we review advances in the understanding of the sarcoidosis granuloma along with contemporary diagnostic and therapeutic considerations for CS. Additionally, we highlight knowledge gaps and areas for future research in CS treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 1899, the dermatologist Caesar Boeck was the first to link “sarcoma giant cells” with a multi-organ disorder comprised of skin findings and lymphadenopathy [1]. More than a century after Boeck’s description, the non-caseating granuloma has become the defining feature of sarcoidosis. However, the granuloma remains a mysterious entity, both in terms of its pathobiology and its clinical manifestations. Imaging modalities that identify scarring and inflammation, as a surrogate for an active granuloma, have led to increased disease recognition and diagnosis. Meanwhile, treatment approaches have focused on attenuating granulomatous inflammation.
The prevalence of sarcoidosis is estimated to be 0.10–0.16% among the US population, with a nearly 3.5-fold increased incidence in African Americans compared to Whites [2, 3]. Although only about 5% of patients with systemic sarcoidosis have clinically manifest cardiac involvement, the true prevalence of cardiac sarcoidosis (CS) is likely underestimated. For example, autopsy studies suggest that cardiac involvement occurs in up to 25% of patients with sarcoidosis [4]. Because patients with sarcoidosis are at a higher risk of death when they exhibit heart failure, atrioventricular block, and ventricular tachycardia, the presence of cardiac involvement is generally accepted to be an indication for treatment [5••, 6••].
Granulomagenesis
A Recipe for Sarcoidosis: Innate Susceptibility and an Environmental Trigger
How and why sarcoidosis develops in some individuals remains largely unknown. However, two key features have emerged: innate susceptibility to sarcoidosis and the need for a triggering event. Functional evidence for this “recipe” is best demonstrated by the Kveim-Siltzbach test, in which sterilized splenic extracts from an individual with sarcoidosis are injected intradermally and followed for the development of granuloma. The Kveim test has been reported to have a sensitivity of 84%, compared to a 1–2% false positive rate [7]. Thus, acellular splenic extracts (exposure) from an individual with sarcoidosis are sufficient to instruct granuloma formation only in individuals with sarcoidosis (susceptibility). Twin studies and genome-wide association studies support a genetic basis for disease susceptibility, with variants of MHC Class II region genes, BTNL2, ANXA11, and NOTCH4 associated with sarcoidosis [8–10]. The second component of sarcoidosis is an inciting environmental trigger. For example, clusters of sarcoidosis, such as among 9/11 first responders, have been identified following mass exposures to dust, composed largely of calcite, gypsum, bassanite, and crystalline silica [11]. The triggering antigen, if any, is likely to vary according to race or ethnic group, geographic location, and genetic background [12]. Some potential offenders include infectious agents such as Mycobacterium and Propionibacterium, organic triggers, and inorganic triggers such as metal exposures to beryllium, zirconium, and aluminum [13].
Cellular Make-up of the Sarcoid Granuloma
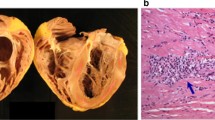
Granulomas, part of an evolutionarily conserved response that arises when monocytes are not fully able to clear an antigen, are typically defined as foci of mature mononuclear phagocytes [14, 15]. The architecture of the granuloma is stereotypical with a central core of macrophages, surrounded by lymphocytes, fibroblasts, blood vessels, and epithelioid cells (Fig. 1). However, the extent of these additional cellular contributions can vary among individuals and even among granulomas. Granulomas are not specific to sarcoidosis and are observed in other conditions including, but not limited to, Mycobacterial infections, fungal infections, Crohn’s disease, vasculitis, foreign body reactions, and inherited genetic disorders such as chronic granulomatous disease [16].

Granuloma-centered approaches to CS treatment. Current sarcoidosis treatments target antigen presentation, T-cell activation, inflammatory chemokines, and granuloma formation [17]. APC, antigen-presenting cell; Th1/Th17 effector T-cells; TNF, tumor necrosis factor; IFN, interferon; IL, interleukin
The prevailing mechanism for sarcoidosis is that phagocytosis of an unknown antigen by an antigen-presenting cell triggers a granulomatous program in macrophages along with activation of an exaggerated CD4+ T-helper 1 (TH1) response, thus incorporating both innate and adaptive immune systems. Like other granulomatous disorders, granulomas of sarcoidosis have a central core of monocyte-derived epithelioid cells and giant cells, surrounded by inflammatory lymphocytes. However, unlike other granulomatous disorders, the central core of the sarcoid granuloma is not necrotic and thought to be sterile. Mechanisms of granulomagenesis for sarcoidosis are largely based on association studies using clinical specimens and lessons learned from other granulomatous disorders.
The different cells involved in granuloma formation are the following:
-
T-cells. Several observations have implicated CD4+ T-cells as critical mediators of sarcoidosis, including the observation that sarcoidosis can go into remission in patients with HIV infection and can manifest during immune reconstitution after the treatment of HIV/AIDS [18–20]. Further supporting a role for CD4+ T-cells are analyses of bronchoalveolar lavage (BAL) samples from individuals with sarcoidosis demonstrating an enrichment of CD4+ TH1 and TH17 T-cells [21]. Granuloma tissue also shows upregulation of the TH1 cytokines IFN-γ, TNF-α, IL-2, IL-12, and IL-18 [12, 21, 22]. Thus, like Mycobacterial granulomas, the sarcoid granuloma is canonically associated with type I immune signaling. In addition to TH1 T-cells, studies on BAL samples suggest that specialized Treg cells, which serve to dampen the adaptive immune response, are relatively depleted or ineffective in patients with sarcoidosis [23, 24].
-
Macrophages. Macrophages and their derivatives are the defining feature of the granuloma. Through single cell hanging drop experiments and genetic lineage tracing, macrophages have been shown to be the cellular source of multinucleated giant cells and epithelioid cells [25, 26]. Interestingly, although both Mycobacterial and sarcoid granulomas are archetypically type I cytokine-mediated structures, recent work in a zebrafish Mycobacterial model has implicated type 2 cytokine signaling via IL4 and STAT6 as the key mediators of the transition of macrophages to epithelioid cells [27]. Importantly, interference with epithelioid transformation by blockade of epithelioid junction proteins or STAT6 signaling promotes pathogen clearance and can even prevent granuloma formation [26, 27]. Additionally, both M1 and M2 macrophage responses have been implicated in CS: the pro-inflammatory M1-type is associated with granuloma formation and the anti-inflammatory M2-type is associated with angiogenesis and a TH2 response. M2-type macrophages can also influence early granuloma formation and promote fibrosis by recruiting and activating fibroblasts to make collagen [28].
-
Other cell types. In addition to T-cells and macrophages, the granulomas of sarcoidosis incorporate other cell types including B-cells, mast cells, and fibroblasts. B-cells tend to be localized in the outer layer of the granuloma and circulating levels of B-cell activating factor (BAFF) are increased in patients with sarcoidosis. Although their role in CS remains to be understood, mast cells may be a modifier of disease severity, as they can produce TNF‐α that activates CD8+ T-cells; are more numerous in patients with high inflammatory activity; and are associated with a more severe disease course [29]. Lastly, fibroblasts are mediators of tissue fibrosis and sources of cytokines, like IL-6, that can enhance the inflammatory response [30, 31].
Cardiac Sarcoidosis
Cardiac Manifestations
Clinical phenotypes of CS can be a result of active inflammation or sequelae of fibrosis [32]. The two most common presentations of CS are arrhythmia, including both brady-arrhythmias and tachyarrhythmia, and heart failure, with either reduced ventricular function or preserved ventricular function. Unlike other infiltrative disorders of the myocardium, sarcoidosis results in patchy granulomatous infiltration and clinical presentations largely follow the location and extent of CS within the heart. While sarcoidosis can affect nearly any region of the heart, there appears to be a tropism for the basal septum and right ventricular myocardium for unclear reasons [33]. This preference for the basal septum correlates with the heart block seen in about 30–40% of individuals with CS [34, 35]. One of the goals of treatment in CS is to limit the amount of fibrosis and tissue loss. However, at present, high-quality evidence is lacking to demonstrate that early treatment of sarcoidosis improves outcomes [32, 36, 37].
Diagnosing CS
Diagnostic Criteria
As a result of increasing recognition of CS, several societies have developed diagnostic criteria that include clinical and histologic elements. The first guidelines were released by the Japanese Ministry of Health and Welfare in 1993 and have since been updated by the Japanese Society of Sarcoidosis and Other Granulomatous Disorders in 2007 and the Japanese Circulation Society (JCS) in 2016 [32, 38, 39]. The 2016 JCS guidelines require either (1) a histologic diagnosis guided by the presence of non-caseating granulomas on endomyocardial biopsy in an individual with clinical features of sarcoidosis or (2) a clinical diagnosis established by the presence of 2 major criteria or 1 major and 2 minor criteria along with a histologic or clinical diagnosis of extracardiac sarcoidosis (Table 1, part A) [6••, 32]. Diagnostic guidelines have also been developed by the Heart Rhythm Society (HRS) that similarly requires histologic and clinical criteria (Table 1, part B) [6••]. For both sets of guidelines, advances and wider prevalence of imaging techniques that can identify cardiac inflammation have led to the use of clinical criterion without the need for confirmatory histology.
To help unify the HRS and the 2007 Japanese guidelines and increase diagnosis of CS, the World Association of Sarcoidosis and Other Granulomatous Diseases (WASOG) created an organ assessment tool [40]. The goal of this tool is to identify the likelihood of organ involvement as highly probable (> 90% likelihood) based on biopsy with presence of granulomas; probable (50–90% likelihood) based on HRS criteria (Table 1, part B); and possible (< 50% likelihood) in those with reduced left ventricular ejection fraction (LVEF) in the absence of other risk factors and in those with atrial dysrhythmias [40].
While these diagnostic criteria have greatly enhanced the recognition of CS, considerable limitations remain. First, requiring the presence of extracardiac disease disregards individuals with isolated CS [41]. To date, the true prevalence of isolated CS is unknown given the prior requirements for histological evidence of extracardiac CS for diagnosis; however, an autopsy study of 46 army patients with sudden cardiac death and known or suspected CS suggest a rate of isolated CS of 29% [33]. To help identify individuals with isolated CS, the JCS 2016 guidelines allow for a clinical diagnosis of isolated CS based on imaging findings and at least three major criteria (Table 1, part A) [42••]. Applying the 2016 JCS guidelines to a cohort of 94 patients with suspected CS led to a diagnosis of isolated CS in 20% of cases. However, diagnoses based on imaging alone may lose specificity for CS. Finally, different diagnostic criteria are not always in agreement. In fact, the 2016 JCS guidelines had a 1.5-fold higher diagnostic yield for diagnosis of CS, with a sensitivity of 81% and specificity of 87% when compared to the HRS and WASOG guidelines [42••].
Clinical Criteria
In general, CS should be considered in younger patients with unexplained arrhythmias and in patients with heart failure with scar patterns inconsistent with coronary heart disease. A study of 351 cases of CS revealed high-grade atrioventricular block was the most common first sign of CS present in 42% of cases, followed by heart failure in 17% of cases, sudden cardiac death in 14% of cases, and sustained ventricular tachycardia in 14% cases [43].
For individuals with suspected CS, current guidelines recommend that a cardiac history, electrocardiogram (ECG), and echocardiogram be performed in patients with extracardiac sarcoidosis in order to screen for cardiac involvement. In those with positive symptoms and abnormal ECG and/or abnormal echocardiogram, advanced cardiac imaging with cMRI and/or cardiac PET is recommended [6••]. This recommendation is based on expert opinion and is likely to underestimate occult cardiac involvement due to the insensitivity of electrocardiography and echocardiography for CS. Holter testing may be another cardiac test to help risk stratify individuals with sarcoidosis for arrhythmic events. Data from a CS multicenter international registry of 587 patients with CS showed Holter monitoring can identify high-risk features such as atrial fibrillation/atrial flutter, atrioventricular block, premature ventricular contraction (PVC) burden, and ventricular tachycardia [44].
Histologic Criteria
In the presence of a compatible clinical syndrome, histologic demonstration of non-caseating granulomas in the myocardium is the only way to definitively diagnose CS. Thus, in patients with a compatible presentation and cardiac magnetic resonance imaging (cMRI) or position emission tomography (PET) findings consistent with CS, right ventricular endomyocardial biopsy is often considered. It is key to differentiate the non-caseating sarcoid granuloma from the caseating granulomas seen in tuberculosis and the giant cells with active necrosis seen in giant cell myocarditis. However, due to the patchy nature of myocardial involvement by sarcoidosis, sampling error from right ventricular endomyocardial biopsies is common with diagnostic yield ranging from ~ 20 to 30% in cases of suspected CS [45, 46]. Given the low sensitivity of endomyocardial biopsy, a diagnosis of CS cannot be ruled out with a negative biopsy result. Thus, for many patients, the risk/benefit ratio of endomyocardial biopsy to diagnose CS may not be favorable and biopsy may not be advised. Different strategies have been proposed to help increase biopsy sensitivity and diagnostic yield. Electro-anatomical mapping of the right and left ventricle may increase the sensitivity of endomyocardial biopsy for sarcoidosis by enabling the sampling of abnormal myocardium. A study of 79 patients with suspected CS who underwent electrogram-guided (EGM) endomyocardial biopsy showed abnormal EGM at the biopsy site had a sensitivity of 89% and specificity of 33% for a histopathologic diagnosis of CS when compared to late gadolinium enhancement on cardiac MRI and cardiac PET [47]. While EGM-guidance may increase biopsy yield, additional work is needed to determine whether EGM-guided biopsy has enough sensitivity to warrant routine implementation.
Imaging Criteria
Cardiac MRI with gadolinium contrast is an important imaging modality for the diagnosis of CS. Gadolinium is an extracellular contrast agent with slow washout from areas of fibrosis and inflammation, enabling identification of sarcoidosis based on scar pattern [48]. CS classically presents with a patchy LGE appearance but can also present with a variety of LGE patterns involving the subendocardial, transmural, mid-wall, and epicardial segments [49]. LGE involvement of the base, mid-ventricle, and subendocardial involvement of the right ventricular side of the septum may increase the specificity for CS [41, 50••]. Cardiac MRI can be also useful for staging. The acute inflammatory stage of CS is marked by myocardial thickening with wall motion abnormalities, increased T2-weighted signal suggestive of edema, and LGE. In the chronic stage, the prevalent findings are focal areas of myocardial thinning with LGE, most common in the basal septum although it can be found in other areas. These areas of LGE in the chronic stage represent scar and/or chronic granulomatous tissue rather than inflammation [Nadel, 2016], [50••]. Cardiac MRI can also be used to differentiate CS from other arrhythmogenic cardiomyopathies, with mediastinal lymphadenopathy and left ventricular septal involvement being more common in the former [51]. However, there are absolute and relative contraindications to cMRI such as allergy to gadolinium contrast, presence of non-MRI compatible implants or foreign bodies, and renal insufficiency that limit the use of cMRI [52].
In addition to cMRI, the use of fluorodeoxyglucose PET (FDG-PET) can be valuable to diagnose and guide the treatment of CS. Inflammatory lesions have a high rate of metabolic activity and glucose utilization that results in increased FDG uptake. Some limitations of this modality include exposure to ionizing radiation, the need for a specialized patient preparation consisting of low carbohydrate meals followed by fasting, and the inability to differentiate inflammation from non-inflammatory myocardial uptake, especially in insulin-dependent diabetics [53, 54]. However, a meta-analysis looking at the role of FDG-PET in diagnosis of CS showed 89% (CI 79–96%) sensitivity and 78% (CI 68–86%) specificity compared to the Japanese Ministry of Health, Labour, and Welfare (MHLW) guidelines diagnostic criteria [54]. Data from a recent meta-analysis of 33 studies comparing the diagnostic accuracy of cMRI versus FDG-PET suggested cMRI had higher sensitivity than FDG-PET (95% vs 84%; P = 0.002), with no difference in specificity (85% vs 82%; P = 0.85) [55]. However, rather than being an either/or set of modalities, cMRI and FDG-PET may be synergistic. Such an approach provides a pathway for detection of active CS among patients with no LGE but abnormal uptake on PET and abnormal T1/T2 mapping [56]. A simultaneous approach with FDG uptake co-localizing with LGE on cMRI has been shown to help predict major adverse cardiac events in CS compared to FDG uptake or LGE alone (HR 14.9, P = 0.001) [57].
In recent years, imaging modalities have become a useful adjunct to titrate immunosuppressive therapy. FDG-PET-guided treatment may enable steroid weaning in patients without evidence of active disease at 1 year of therapy [Ning, 2019]. Moreover, a myocardial FDG uptake index > 30 and LVEF > 40% on pre-treatment FDG-PET has been shown to be a predictor of clinical and electrocardiographic response to treatment (HR 1.28 and 1.61, respectively) and had a correlation with change in LVEF after immunosuppression [58]. The baseline maximum standardized uptake value (SUVmax) and degree of metabolic response on FDG-PET after treatment with steroids have also been shown to help distinguish patients with higher risk of relapse and with worse prognosis [58]. Similar to FDG uptake, changes in myocardial T2 signals before and after treatment can be used to assay for active inflammation and guide therapy [59, 60]. The optimal timing on the use of FDG-PET and/or cMRI to help guide response to therapy, predict relapse, and guide medication titration or device implantation remains to be determined.
Serum Markers
Although several biomarkers have been proposed, no single serum marker is specific or sensitive enough to help with diagnosis and/or to guide response to therapy in CS. For example, serum angiotensin-converting enzyme (ACE) levels are elevated in 60% of patients with sarcoidosis but are also frequently elevated in other inflammatory states [61, 62]. Furthermore, ACE levels cannot be used to aid diagnosis or disease monitoring in individuals taking concurrent ACE inhibitors [63]. More recently, other inflammatory markers such as the soluble interleukin 2 receptor (sIL-2R) which is made by activated T-cells and serum adenosine deaminase 2 (ADA-2) levels have been proposed as inflammatory biomarkers of diagnostic and prognostic significance for sarcoidosis [64, 65].
In addition to inflammatory biomarkers, cardiac markers have been used to follow CS. NT-proBNP is a marker of cardiac stress and has been shown to be elevated in patients with CS [Handa, 2010]. High-sensitivity cardiac troponin (hs-cTn), released with myocardial injury, may be comparable to BNP in terms of sensitivity (87.5% vs 87.5%) and superior in terms of specificity (75% vs 50%) [66]. Moreover, hs-cTn has the potential to be used to follow treatment response in patients with CS. Prior work showed that up to 67% of patients with new-onset CS had elevated hs-cTn levels, which normalized after 4 weeks of steroid therapy [67]. However, the value of serial hs-cTn and/or NT-pro BNP levels has yet to be determined.
Treatment of CS: A Focus on the Granuloma
Immunosuppression
Current recommendations for treatment are largely based often on single-center experiences and/or consensus opinion [6••] [Supplementary Material]. The decision to treat with systemic immunosuppression is often personalized based on severity of disease and patient-risk. The HRS guidelines recommend starting immunosuppression in patients with active CS as evidenced by findings of active inflammation on cMRI and/or positive uptake on FDG-PET along with any of the following: high-degree AV block, frequent ventricular ectopy, and non-sustained or sustained ventricular arrhythmias [6••]. To date, there is no clear consensus on whether to treat asymptomatic individuals with CS and positive imaging findings on CMR and/or FDG-PET. However, based on what is known about the pathophysiology of CS, a goal of treatment is to prevent potentially irreversible myocardial fibrosis and adverse LV remodeling that can lead to intractable arrhythmias and heart failure [68].
Steroids
Steroids are considered the first-line treatment in patients with active CS based on expert consensus [6••, 69]. Steroids reduce inflammation and may attenuate granuloma formation [70]. Observational datasets suggest a benefit to the use of steroids, and response to steroids is a diagnostic criterion in the HRS guidelines [6••]. Treatment of active sarcoidosis with glucocorticoids has been associated with less ventricular dysfunction over time [71–75]. Even in individuals with severe LV dysfunction, steroids have been associated with recovery of ventricular function [67]. Steroid effects on arrhythmia are more mixed, with a likely benefit in the treatment of AV conduction disorders, but less prominent effects on ventricular arrhythmias [69, 76–78].
Many questions remain on steroid dosing and duration of therapy to prevent long-term side effects. An ongoing CS Randomized Trial (CHASM-CS-RCT) was designed to help answer this question. The results of this trial are expected within the next 4 years and will likely provide more clarity on the initial approach to treatment of CS [79••].
Steroid-Sparing Agents
Because long-term steroid use is associated with numerous adverse effects, such as hypertension, diabetes, weight gain, osteoporosis, and increased risk of infections, steroid-sparing agents are being increasingly used for CS. Steroid-sparing agents may be considered adjunctive measures to limit steroid dosing in CS and improve treatment responses. The combination of steroids and immunosuppressive drugs at the time of CS diagnosis might reduce the risk of CS relapse compared to steroids alone, with relapse rates of 45.8% in the steroid group versus 16.7% in the steroids and immunosuppression group (P = 0.048) [80].
Methotrexate (MTX) is the most widely studied steroid-sparing agent for CS. The mechanism of action of methotrexate is thought to be antimetabolite formation in T-helper cells [81]. Methotrexate along with corticosteroids has been associated with lower rates of CS relapse compared to treatment with steroids alone. Relapse in this study was defined by several surrogate endpoints, including LVEF, BNP levels, and cardiothoracic ratio on chest radiograph [5••, 73].
Mycophenolate mofetil (MMF), a prodrug of mycophenolic acid (MPA), is another antimetabolite that has been studied as steroid-sparing therapy for CS. MPA depletes guanosine nucleotides preferentially in T and B lymphocytes and inhibits their proliferation, further suppressing adaptive immune responses and antibody formation. MMF use was associated with improved survival on univariate analysis from a single-center cohort of 73 patients with CS, 10 out of which were treated with MMF [82]. More recently, a retrospective study of 77 patients with CS treated with prednisone (n = 32) or prednisone plus MMF (n = 45) showed MMF therapy was well tolerated, and that combination therapy led to a lower total exposure to maximum prednisone dose (P = 0.06) [83••].
Azathioprine is an inhibitor of purine synthesis that leads to inhibition of T- and B-cell proliferation and cytotoxic T-cell function [84, 85]. To date, there are no large prospective studies looking at the use of single steroid-sparing agents on CS treatment. Data is extrapolated from small retrospective studies. In one such analysis, 12 patients receiving steroid-sparing treatment with methotrexate (n = 5), azathioprine (n = 5), or cyclophosphamide (n = 2) were compared to 24 patients receiving steroid-only therapy over a median follow-up of 3.6 months. Clinical relapse (defined as LVEF reduction, 3rd degree atrioventricular block, atrial/ventricular tachycardia, or sudden cardiac death) was seen in 16.7% of patients in the combination therapy group versus in 45.8% of patients in the steroid-only group (P = 0.048) and there was a tendency toward lower relapse rates in the combination therapy group compared to the steroid-only group (HR = 2.96; 95% CI 0.66–13.48; log-rank, P = 0.141) [80]. Although hypothesis generating, currently available studies have many limitations including small sample size, retrospective approach, short follow-up time, and different disease severities among treatment groups.
Biologics
TNF-α inhibitors, which directly antagonize the TH1 cellular response, have been increasingly used in CS. Most studies have focused on infliximab, a chimeric monoclonal antibody which binds circulating TNF-α, and adalimumab, a fully human monoclonal antibody that inhibits TNF-α binding to its receptor. Anti-TNF agents are controversial in patients with heart failure, based on prior trial results. Infliximab was evaluated in the phase II clinical trial Anti-TNF-α Therapy Against Chronic Heart Failure (ATTACH) that randomized patients with stable NYHA III-IV and LVEF of ≤ 35% to either infliximab or placebo. Infliximab was associated with a dose-related increase in death and heart failure hospitalizations when compared to placebo. Of note, adverse outcomes were seen only with the highest dose of infliximab (10 mg/kg) which was associated with an increase in all-cause mortality and number of HF hospitalizations [86].
Recent work has challenged the extrapolation of safety concerns for anti-TNF agent use from patients with heart failure to patients with CS. For example, in a cohort of 20 patients receiving TNF-α inhibitors, none had worsening heart failure, and all had clinical benefit. Out of the patients treated with TNF-α inhibitors, repeat imaging showed resolution of disease activity [87]. Another retrospective study of 16 patients with CS and refractory dysrhythmias, moderate to severe cardiomyopathy, and persistent FDG uptake on PET showed treatment with infliximab led to a reduction in prednisone-equivalent steroid doses from a median of 20 to 5 mg at 12 months post-initiation of infliximab (P < 0.001). Additionally, there was a trend toward reduction of ventricular tachycardia events from 32 at baseline to 19% at 12 months (P = 0.07) [88]. Despite concerns that TNF-α inhibitors can worsen heart failure, there were no patients with a notable decline in LVEF at 1 year, and most patients saw an improvement. Another multicenter study of TNF-α inhibitor use in 38 patients with CS, guided by FDG-PET response to treatment, showed reduced corticosteroid use and reduced cardiac inflammation without significant adverse effect on cardiac function with TNF-α inhibitors [89]. A recent retrospective cohort of 9 patients with metabolically active CS (based on PET imaging) showed that 6 of the 9 patients had complete resolution of active CS and no change in LVEF during the follow-up period [90••]. The body of evidence on the use of TNF-α inhibitors continues to grow and a multicenter randomized trial is clearly needed to definitively address the question of safety and efficacy of anti-TNF agents in patients with CS.
The story on immunosuppressive therapy continues to unfold and other agents on the frontier for CS treatment include rituximab, a chimeric monoclonal antibody against the protein CD20, primarily found on B-cells. Rituximab has been used in the management of CS cases that have failed treatment with steroids ± steroid-sparing agents. A recent review of a single-center use of rituximab in 7 patients with active CS showed a reduction in inflammation as measured by cardiac FDG PET/CT uptake in 6 of 7 patients with LVEF improvements or stabilization in 4 patients and a decrease in 3 [91].
Concurrent Management of Cardiovascular Disorders
Along with the immunosuppression, patients with CS should be treated with guideline-directed medical therapy (GDMT) for heart failure. The scope of the heart failure guidelines will not be discussed in this review but patients with CS should be considered to be American Heart Association (AHA) stage B in the absence of overt heart failure and AHA stage C/D disease depending on the severity of heart failure [92].
In addition to GDMT, concurrent management of cardiovascular disorders includes arrhythmia management. Antiarrhythmic therapy should include beta-blockade and/or drugs that prolong repolarization such as amiodarone and sotalol. The use of class IA antiarrhythmic drugs is not recommended in patients with low LVEF [93••]. A key question is whether the arrhythmogenesis in CS is mediated by inflammation or scar. Imaging by PET-CT or cardiac MRI can be instructive and is recommended. Persistent inflammation despite immunosuppression should prompt either addition of a second agent or consideration of transplantation. For scar-mediated ventricular arrhythmias, catheter ablation is an option to consider in select patients with CS. A multicenter analysis looking at patients with CS undergoing VT catheter ablation showed that antiarrhythmic drug requirements and defibrillator shocks were reduced, and VT storm was eliminated in 82% of patients with CS over a follow-up of 2.5 years. In this study, pre-ablation EF < 50% was associated with worse long-term prognosis [94].
Lastly, an important consideration in the management of CS is the use of intracardiac defibrillators (ICD) to prevent sudden cardiac death. While low LVEF is a strong risk factor for ventricular arrhythmias and sudden cardiac death in CS, those with normal or near-normal LVEF also carry substantial risk. For CS patients with an indication for permanent pacing, there is a class IIa recommendation for concurrent ICD placement in those with a history of unexplained syncope or extensive scar by cardiac MRI or PET, but the extent of scar considered for ICD recommendation is not yet known [93••]. In diseases such as hypertrophic cardiomyopathy (HCM), LGE burden > 15% doubles the risk of sudden cardiac death, although recent studies suggest patients with HCM and > 5% LGE should be carefully considered for ICD implantation [95]. Moreover, CS is an inflammatory condition with a different pathophysiology to HCM and applying similar cutoffs might underestimate arrhythmia risk among individuals with a lower burden of LGE.
Outside of those with an indication for pacing, which CS patients with normal ventricular function should be considered for primary prevention ICDs has not been determined [96]. The ACC/AHA/HRS guidelines give a class I recommendation for primary prevention ICD in individuals with CS who have inducible ventricular arrhythmias on EP study [6••, 93••]. However, which patients might qualify for EP study is not well defined. Alternatively, ambulatory event monitoring might help risk stratify patients for primary prevention ICDs [44].
Advanced Heart Failure Therapies
As the identification of CS increases, the number of patients needing advanced heart failure therapies such as left ventricular assist devices (LVAD) and/or cardiac transplantation continues to increase. Heart transplantation is an increasingly common option in CS patients with end-stage heart failure or refractory ventricular arrhythmias. There are important considerations to keep in mind when evaluating CS patients for LVAD or heart transplantation and these include the degree of extracardiac involvement and the degree of RV involvement. Moreover, LVAD therapy is not considered a therapeutic option in patients with refractory ventricular arrhythmias as LVAD support may not prevent arrhythmia burden and may be less effective with persistent arrhythmia [97]. However, CS is not an absolute contraindication for LVAD support, as carefully selected patients with CS who underwent LVAD implantation as a bridge to heart transplantation can have similar 1- and 5-year survival to other patients with non-ischemic cardiomyopathies [98].
A recent study of 227 patients with CS undergoing heart transplantation showed similar post-transplantation outcomes in terms of survival, post-transplant malignancy, odds of graft failure, and infection as patients without cardiac sarcoidosis [99••]. Another recent study showed that short-term and long-term outcomes after heart transplantation are similar among patients with CS compared to those with other types of non-ischemic cardiomyopathies [100]. Although there is a small risk of CS recurrence post-transplant and donor to recipient transmission, there are only case reports of this happening, and it has been suggested that contemporary immunosuppression regimens used for transplant recipients may limit progression of sarcoidosis [100, 101]. Overall, outcomes after transplantation for CS are favorable.
Discussion
CS is a dynamic field, with increasing disease recognition of affected patients and new diagnostic and treatment modalities on the horizon. However, key questions remain as to the optimal approaches for diagnosing, following, and treating CS. In the absence of multicenter, randomized clinical trials, guidelines are largely based on expert opinion.
Our center relies heavily on imaging for the diagnosis of CS (both cMRI and FDG-PET). We strongly pursue a histologic confirmation of sarcoidosis to increase the probability that imaging patterns are specific for sarcoidosis. In cases without apparent extracardiac disease and when imaging predicts involvement of the right ventricular septum, we pursue endomyocardial biopsy to confirm the diagnosis of CS. Additionally, we use T2 signal and changes in scar burden on cardiac MRI, changes in FDG uptake, changes in systolic function, and arrhythmia burden on ICD interrogation to determine treatment responses.
Our decision to treat is based on the probability for cardiac sarcoidosis and patient-level risk. We use a similar probabilistic approach to CS diagnosis as the one used by WASOG and HRS guidelines [6••, 40]. For possible CS and low-risk features, we clinically observe these patients for symptoms or progression of imaging findings. For patients with possible CS and high-risk features, or probable/definite CS, we treat. Our initial treatment generally consists of 0.5 mg/kg prednisone daily (max dose 40 mg) followed by imaging to assess for disease progression at 3 months. If there is evidence of active disease on repeat imaging, we escalate therapy to include a steroid-sparing patient and/or an anti-TNF agent. In cases with isolated CS and disease refractory to prednisone, we pursue voltage-guided biopsy to verify a diagnosis of CS. For patients that respond to prednisone, we transition to a steroid-sparing regiment for at least 1 year with interval imaging to confirm disease quiescence. After 1 year, we use a shared decision-making approach to determine whether to continue or discontinue immunosuppression based on surveillance imaging. In addition to immunosuppression, we treat systolic dysfunction and arrhythmias based on the guidelines cited earlier. Finally, and perhaps most importantly, we utilize a multidisciplinary approach with heart failure cardiologists, electrophysiologists, radiologists, and rheumatologists to make individualized treatment decisions.
While most centers utilize a similar approach, the lack of data driving the management of sarcoidosis needs to be acknowledged and addressed.
Conclusion
This review illustrates how many key questions around how to diagnose CS, which patients to treat, what the optimal approach to treatment is, how to assess treatment responses, and when to discontinue treatment remain unanswered. Moving beyond single-center retrospective studies to pragmatic, multicenter randomized studies will be critical to advance the field with high-quality data. Doing so will help elucidate which treatments are consistently beneficial across different centers and patient populations.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Young RC Jr, Rachal RE, Cowan CL Jr. Sarcoidosis—the beginning: historical highlights of personalities and their accomplishments during the early years. J Natl Med Assoc. 1984;76(9):887.
Rybicki BA, Major M, Popovich J Jr, Maliank MJ, lannuzzi MC. Racial differences in sarcoidosis incidence: a 5-year study in a health maintenance organization. Am J Epidemiol. 1997;145(3):234–41.
Arkema EV, Cozier YC. Epidemiology of sarcoidosis: current findings and future directions. Ther Adv Chronic Dis. 2018;9(11):227–40.
Iwai K, Tachibana T, Takemura T, Matsui Y, Kitalchi M, Kawabata Y. Pathological studies on sarcoidosis autopsy. I. Epidemiological features of 320 cases in Japan. Pathol Int. 1993;43(7–8):372–6.
•• Rosenthal DG, Parwani P, Murray TO, Petek BJ, Benn BS, De Marco T, et al. Long-term corticosteroid-sparing immunosuppression for cardiac sarcoidosis. J Am Heart Assoc. 2019;8(18):e010952. Study showing steroid-sparing agents are an effective maintenance therapy in patients with cardiac sarcoidosis after an initial response to steroids is achieved.
•• Birnie DH, Sauer WH, Bogun F, Cooper JM, Culver DA, Duvernoy CS, et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm. 2014;11(7):1304–23. Most recent guidelines on diagnosis and treatment of cardiac sarcoidosis.
Siltzbach LE. The Kveim test in sarcoidosis: a study of 750 patients. JAMA. 1961;178(5):476–82.
Adrianto I, Lin CP, Hale JJ, Levin AM, Datta I, Parker R, et al. Genome-wide association study of African and European Americans implicates multiple shared and ethnic specific loci in sarcoidosis susceptibility. PLoS One. 2012;7(8):e43907.
Valentonyte R, Hampe J, Huse K, Rosenstiel P, Albrecht M, Stenzel A, et al. Sarcoidosis is associated with a truncating splice site mutation in BTNL2. Nat Genet. 2005;37(4):357–64.
Hofmann S, Franke A, Fischer A, Jacobs G, Nothnagel M, Gaede KI, et al. Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat Genet. 2008;40(9):1103–6.
Izbicki G, Chavko R, Banauch GI, Weiden MD, Berger KI, Aldrich TK, et al. World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest. 2007;131(5):1414–23.
Drent M, Crouser ED, Grunewald J. Challenges of sarcoidosis and its management. N Engl J Med. 2021;385(11):1018–32.
Birnie DH, Nery PB, Ha AC, Beanlands RSB. Cardiac sarcoidosis. J Am Coll Cardiol. 2016;68(4):411–21.
Pagán AJ, Ramakrishnan L. The formation and function of granulomas. Annu Rev Immunol. 2018;36:639–65.
Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity. 2002;17(6):693–702.
James DG, Zumla A. The granulomatous disorders. Cambridge: Cambridge Univ. Press; 1999. p. 132–60.
Gerke AK. Treatment of sarcoidosis: a multidisciplinary approach. Front Immunol. 2020;11:545413.
Vega LE, Espinoza LR. HIV infection and its effects on the development of autoimmune disorders. Pharmacol Res. 2018;129:1–9.
Foulon G, Wislez M, Naccache J, Blanc F, Rabbat A, Dominique I, et al. Sarcoidosis in HIV-infected patients in the era of highly active antiretroviral therapy. Clin Infect Dis. 2004;38(3):418–25.
Morris DG, Jasmer RM, Huang L, Gotway MB, Nishimura S, King TE Jr. Sarcoidosis following HIV infection: evidence for CD4 lymphocyte dependence. Chest. 2003;124(3):929–35.
Ramstein J, Broos CE, Simpson LJ, Ansel KM, Sun SA, Ho ME, et al. IFN-γ–producing T-helper 17.1 cells are increased in sarcoidosis and are more prevalent than T-helper type 1 cells. Am J Respir Crit Care Med. 2016;193(11):1281–91.
Facco M, Cabrelle A, Teramo A, Olivieri V, Gnoato M, Teolato S, et al. Sarcoidosis is a Th1/Th17 multisystem disorder. Thorax. 2011;66(2):144–50.
Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, et al. The immune paradox of sarcoidosis and regulatory T cells. J Exp Med. 2006;203(2):359–70.
Taflin C, Miyara M, Nochy D, Valeyre D, Naccache J, Altare F, et al. FoxP3 regulatory T cells suppress early stages of granuloma formation but have little impact on sarcoidosis lesions. Am J Pathol. 2009;174(2):497–508.
Cronan MR, Beerman RW, Rosenberg AF, Saelens JW, Johnson MG, Oehlers SH, et al. Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity. 2016;45(4):861–76.
Adams DO. The structure of mononuclear phagocytes differentiating in vivo: I. Sequential fine and histologic studies of the effect of Bacillus Calmette-Guerin (BCG). Am J Pathol. 1974;76(1):17.
Cronan MR, Hughes EJ, Brewer WJ, Viswanathan G, Hunt EG, Singh B, et al. A non-canonical type 2 immune response coordinates tuberculous granuloma formation and epithelialization. Cell. 2021;184(7):1757–74 (e14).
Giblin GT, Murphy L, Stewart GC, Desai AS, Di Carli MF, Blankstein R, et al. Cardiac sarcoidosis: when and how to treat inflammation. Card Fail Rev. 2021 Mar;7.
Kullberg S, Rivera NV, Abo Al Hayja M, Grunewald J, Eklund A. Changes in lung immune cells related to clinical outcome during treatment with infliximab for sarcoidosis. Clin Exp Immunol. 2020;201(1):85–93.
Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657.
Tamura R, Sato A, Chida K, Suganuma H. Fibroblasts as target and effector cells in Japanese patients with sarcoidosis. Lung. 1998;176(2):75–87.
Terasaki F, Yoshinaga K. New guidelines for diagnosis of cardiac sarcoidosis in Japan. Ann Nucl Cardiol. 2017;3(1):42–5.
Tavora F, Cresswell N, Li L, Ripple M, Solomon C, Burke A. Comparison of necropsy findings in patients with sarcoidosis dying suddenly from cardiac sarcoidosis versus dying suddenly from other causes. Am J Cardiol. 2009;104(4):571–7.
Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169(3):510–22 (e20).
Nordenswan H, Lehtonen J, Ekström K, Kandolin R, Simonen P, Mäyränpää M, et al. Outcome of cardiac sarcoidosis presenting with high-grade atrioventricular block. Circ Arrhythm Electrophysiol. 2018;11(8):e006145.
Padala SK, Peaslee S, Sidhu MS, Steckman DA, Judson MA. Impact of early initiation of corticosteroid therapy on cardiac function and rhythm in patients with cardiac sarcoidosis. Int J Cardiol. 2017;227:565–70.
Fussner LA, Karlstedt E, Hodge DO, Fine NM, Kalra S, Carmona EM, et al. Management and outcomes of cardiac sarcoidosis: a 20-year experience in two tertiary care centers. Eur J Heart Fail. 2018;20(12):1713–20.
Hiraga H, Iwai K, Hiroe M, Omori F, Sekiguchi M, Tachibana T. Guidelines for diagnosis of cardiac sarcoidosis: study report on diffuse pulmonary diseases (in Japanese) Tokyo: The Japanese Ministry of Health and Welfare; 1993. Google Scholar :23–24.
Hiraga H, Yuwai K, Hiroe M. Diagnostic standard and guidelines for sarcoidosis. Jpn J Sarcoidosis Granulomatous Disord. 2007;27(89):102.
Judson MA, Costabel U, Drent M, Wells A, Maier L, Koth L, et al. The WASOG sarcoidosis organ assessment instrument: an update of a previous clinical tool. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31(1):19–27.
Blankstein R, Waller AH. Evaluation of known or suspected cardiac sarcoidosis. Circ Cardiovasc Imaging. 2016;9(3):e000867.
•• Kawai H, Sarai M, Kato Y, Naruse H, Watanabe A, Matsuyama T, et al. Diagnosis of isolated cardiac sarcoidosis based on new guidelines. ESC Heart Fail. 2020;7(5):2662–71. This study showed how the most recent criteria for CS led to a higher diagnostic yield for CS, particularly isolated CS.
Ekström K, Lehtonen J, Nordenswan H, Mäyränpää MI, Räisänen-Sokolowski A, Kandolin R, et al. Sudden death in cardiac sarcoidosis: an analysis of nationwide clinical and cause-of-death registries. Eur Heart J. 2019;40(37):3121–8.
Bressi E, Crawford TC, Bogun FM, Gu X, Ellenbogen KA, Chicos AB, et al. Arrhythmia monitoring and outcomes in patients with cardiac sarcoidosis: insights from the cardiac sarcoidosis consortium. J Am Heart Assoc. 2022;11(13):e024924.
Uemura A, Morimoto S, Hiramitsu S, Kato Y, Ito T, Hishida H. Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138(2):299–302.
Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. Eur Heart J. 2007;28(24):3076–93.
Ezzeddine FM, Kapa S, Rosenbaum A, Blauwet L, Deshmukh AJ, AbouEzzeddine OF, et al. Electrogram-guided endomyocardial biopsy yield in patients with suspected cardiac sarcoidosis and relation to outcomes. J Cardiovasc Electrophysiol. 2021;32(9):2486–95.
Mahrholdt H, Goedecke C, Wagner A, Meinhardt G, Athanasiadis A, Vogelsberg H, et al. Cardiovascular magnetic resonance assessment of human myocarditis: a comparison to histology and molecular pathology. Circulation. 2004;109(10):1250–8.
Komada T, Suzuki K, Ishiguchi H, Kawai H, Okumura T, Hirashiki A, et al. Magnetic resonance imaging of cardiac sarcoidosis: an evaluation of the cardiac segments and layers that exhibit late gadolinium enhancement. Nagoya J Med Sci. 2016;78(4):437.
•• Patel MR, Cawley PJ, Heitner JF, Klem I, Parker MA, Jaroudi WA, et al. Detection of myocardial damage in patients with sarcoidosis. Circulation. 2009;120(20):1969–77. This study showed LGE on cMRI was more sensitive than prior consensus criteria for the detection of CS.
Steckman DA, Schneider PM, Schuller JL, Aleong RG, Nguyen DT, Sinagra G, et al. Utility of cardiac magnetic resonance imaging to differentiate cardiac sarcoidosis from arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2012;110(4):575–9.
Dill T. Contraindications to magnetic resonance imaging. Heart. 2008;94(7):943–8.
Bokhari S, Sheikh T. Cardiac sarcoidosis: Advantages and limitations of advanced cardiac imaging. J Nucl Cardiol. 2021:1–4.
Youssef G, Leung E, Mylonas I, Nery P, Williams K, Wisenberg G, et al. The use of 18F-FDG PET in the diagnosis of cardiac sarcoidosis: a systematic review and metaanalysis including the Ontario experience. J Nucl Med. 2012;53(2):241–8.
Aitken M, Chan MV, Urzua Fresno C, Farrell A, Islam N, McInnes MD, et al. Diagnostic accuracy of cardiac MRI versus FDG PET for cardiac sarcoidosis: a systematic review and meta-analysis. Radiology 2022:213170.
Greulich S, Gatidis S, Gräni C, Blankstein R, Glatthaar A, Mezger K, et al. Hybrid cardiac magnetic resonance/fluorodeoxyglucose positron emission tomography to differentiate active from chronic cardiac sarcoidosis. Cardiovasc Imaging. 2022;15(3):445–56.
Cheung E, Ahmad S, Aitken M, Chan R, Iwanochko RM, Balter M, et al. Combined simultaneous FDG-PET/MRI with T1 and T2 mapping as an imaging biomarker for the diagnosis and prognosis of suspected cardiac sarcoidosis. Eur J Hybrid Imaging. 2021;5(1):1–18.
Subramanian M, Swapna N, Ali AZ, Saggu DK, Yalagudri S, Kishore J, et al. Pre-treatment myocardial 18FDG uptake predicts response to immunosuppression in patients with cardiac sarcoidosis. JACC: Cardiovasc Imaging. 2021;14(10):2008–16.
Ramirez R, Trivieri M, Fayad ZA, Ahmadi A, Narula J, Argulian E. Advanced imaging in cardiac sarcoidosis. J Nucl Med. 2019;60(7):892–8.
Orii M, Hirata K, Tanimoto T, Ota S, Shiono Y, Yamano T, et al. Comparison of cardiac MRI and 18F-FDG positron emission tomography manifestations and regional response to corticosteroid therapy in newly diagnosed cardiac sarcoidosis with complete heart block. Heart Rhythm. 2015;12(12):2477–85.
Studdy P, Bird R, James DG, Sherlock S. Serum angiotensin-converting enzyme (SACE) in sarcoidosis and other granulomatous disorders. Lancet. 1978;312(8104):1331–4.
Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357(21):2153–65.
Kraaijvanger R, Janssen Bonás M, Vorselaars AD, Veltkamp M. Biomarkers in the diagnosis and prognosis of sarcoidosis: current use and future prospects. Front Immunol. 2020;11:1443.
Kobayashi Y, Sato T, Nagai T, Hirata K, Tsuneta S, Kato Y, et al. Association of high serum soluble interleukin 2 receptor levels with risk of adverse events in cardiac sarcoidosis. ESC Heart Fail. 2021;8(6):5282–92.
Gungor S, Ozseker F, Yalcinsoy M, Akkaya E, Can G, Eroglu H, et al. Conventional markers in determination of activity of sarcoidosis. Int Immunopharmacol. 2015;25(1):174–9.
Baba Y, Kubo T, Kitaoka H, Okawa M, Yamanaka S, Kawada Y, et al. Usefulness of high-sensitive cardiac troponin T for evaluating the activity of cardiac sarcoidosis. Int Heart J. 2012;53(5):287–92.
Kandolin R, Lehtonen J, Airaksinen J, Vihinen T, Miettinen H, Ylitalo K, et al. Cardiac sarcoidosis: epidemiology, characteristics, and outcome over 25 years in a nationwide study. Circulation. 2015;131(7):624–32.
Kouranos V, Sharma R. Cardiac sarcoidosis: state-of-the-art review. Heart. 2021;107(19):1591–9.
Sadek MM, Yung D, Birnie DH, Beanlands RS, Nery PB. Corticosteroid therapy for cardiac sarcoidosis: a systematic review. Can J Cardiol. 2013;29(9):1034–41.
Milburn HJ, Poulter LW, Dilmec A, Cochrane GM, Kemeny DM. Corticosteroids restore the balance between locally produced Th1 and Th2 cytokines and immunoglobulin isotypes to normal in sarcoid lung. Clin Exp Immunol. 1997;108(1):105–13.
Chiu C, Nakatani S, Zhang G, Tachibana T, Ohmori F, Yamagishi M, et al. Prevention of left ventricular remodeling by long-term corticosteroid therapy in patients with cardiac sarcoidosis. Am J Cardiol. 2005;95(1):143–6.
Yazaki Y, Isobe M, Hiroe M, Morimoto S, Hiramitsu S, Nakano T, et al. Prognostic determinants of long-term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88(9):1006–10.
Nagai T, Nagano N, Sugano Y, Asaumi Y, Aiba T, Kanzaki H, et al. Effect of corticosteroid therapy on long-term clinical outcome and left ventricular function in patients with cardiac sarcoidosis. Circ J. 2015;79(7):1593–600.
Nabeta T, Hara M, Naruke T, Maemura K, Oki T, Yazaki M, et al. Clinical valuables related to resolution of complete or advanced atrioventricular block after steroid therapy in patients with cardiac sarcoidosis. J Arrhythm. 2021;37(4):1093–100.
Kato Y, Morimoto S, Uemura A, Hiramitsu S, Ito T, Hishida H. Efficacy of corticosteroids in sarcoidosis presenting with atrioventricular block. Sarcoidosis Vasc Diffuse Lung Dis Off J WASOG. 2003;20(2):133–7.
Fazelpour S, Sadek MM, Nery PB, Beanlands RS, Tzemos N, Toma M, et al. Corticosteroid and immunosuppressant therapy for cardiac sarcoidosis: a systematic review. J Am Heart Assoc. 2021;10(17):e021183.
Banba K, Kusano KF, Nakamura K, Morita H, Ogawa A, Ohtsuka F, et al. Relationship between arrhythmogenesis and disease activity in cardiac sarcoidosis. Heart Rhythm. 2007;4(10):1292–9.
Yodogawa K, Seino Y, Ohara T, Takayama H, Katoh T, Mizuno K. Effect of corticosteroid therapy on ventricular arrhythmias in patients with cardiac sarcoidosis. Ann Noninvasive Electrocardiol. 2011;16(2):140–7.
•• Birnie D, Beanlands RS, Nery P, Aaron SD, Culver DA, DeKemp RA, et al. Cardiac Sarcoidosis multi-center randomized controlled trial (CHASM CS-RCT). Am Heart J. 2020;220:246–52. The results of this RCT will be available in the upcoming years and will compare steroid use to steroid followed by steroid sparing agents for the treatment of CS.
Ballul T, Borie R, Crestani B, Daugas E, Descamps V, Dieudé P, et al. Treatment of cardiac sarcoidosis: a comparative study of steroids and steroids plus immunosuppressive drugs. Int J Cardiol. 2019;276:208–11.
Friedman B, Cronstein B. Methotrexate mechanism in treatment of rheumatoid arthritis. Joint Bone Spine. 2019;86(3):301–7.
Zhou Y, Lower EE, Li H, Costea A, Attari M, Baughman RP. Cardiac sarcoidosis: the impact of age and implanted devices on survival. Chest. 2017;151(1):139–48.
•• Griffin JM, Chasler J, Wand AL, Okada DR, Smith JN, Saad E, et al. Management of cardiac sarcoidosis using mycophenolate mofetil as a steroid-sparing agent. J Card Fail. 2021;27(12):1348–58. Recent study showing mycophenalate mofetil in combination with prednisone is effective and can lead to less steroid use.
Broen JC, van Laar JM. Mycophenolate mofetil, azathioprine and tacrolimus: mechanisms in rheumatology. Nat Rev Rheumatol. 2020;16(3):167–78.
Lewis SJ, Ainslie GM, Bateman ED. Efficacy of azathioprine as second-line treatment in pulmonary sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis Off J WASOG. 1999;16(1):87–92.
Sarzi-Puttini P, Atzeni F, Shoenfeld Y, Ferraccioli G. TNF-α, rheumatoid arthritis, and heart failure: a rheumatological dilemma. Autoimmun Rev. 2005;4(3):153–61.
Baker MC, Sheth K, Witteles R, et al. TNF-alpha inhibition for the treatment of cardiac sarcoidosis. In: Seminars in arthritis and rheumatism. WB Saunders; 2020 Jun 1;50(3):546–52.
Harper LJ, McCarthy M, Neto MLR, Hachamovitch R, Pearson K, Bonanno B, et al. Infliximab for refractory cardiac sarcoidosis. Am J Cardiol. 2019;124(10):1630–5.
Gilotra NA, Wand AL, Pillarisetty A, Devraj M, Pavlovic N, Ahmed S, et al. Clinical and imaging response to tumor necrosis factor alpha inhibitors in treatment of cardiac sarcoidosis: a multicenter experience. J Card Fail. 2021;27(1):83–91.
•• Cundiff M, Mintz D, Pincus M, Campagna A, Nghiem L, Dou Y, et al. Treatment of Active Cardiac Sarcoidosis with TNF Alpha Inhibitors. J Card Fail. 2019;25(8, Supplement):S27–8. In this study, TNF alpha inhibition lead to improvements in inflammation and minimal adverse effects in patients with CS.
Elwazir M, Krause ML, Bois JP, Christopoulos G, Kendi AT, Cooper JLT, et al. Rituximab for the treatment of refractory cardiac sarcoidosis: a single-center experience. J Card Fail. 2022;28(2):247–58.
Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2022;79(17):e263–421.
•• Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2018;72(14):e91–220. These are the most recent arrhythmia guidelines that provide recommendations on ICD use for primary and secondary prevention in patients with CS.
Siontis KC, Santangeli P, Muser D, Marchlinski FE, Zeppenfeld K, Hoogendoorn JC, et al. Outcomes associated with catheter ablation of ventricular tachycardia in patients with cardiac sarcoidosis. JAMA cardiology. 2022;7(2):175–83.
Greulich S, Seitz A, Herter D, Günther F, Probst S, Bekeredjian R, et al. Long-term risk of sudden cardiac death in hypertrophic cardiomyopathy: a cardiac magnetic resonance outcome study. Eur Heart J Cardiovasc Imaging. 2021;22(7):732–41.
Rosenthal DG, Cheng RK, Petek BJ, Masri SC, Mikacenic C, Raghu G, et al. Risk of adverse cardiovascular events in cardiac sarcoidosis independent of left ventricular function. Am J Cardiol. 2020;127:142–8.
Feldman D, Pamboukian SV, Teuteberg JJ, Birks E, Lietz K, Moore SA, et al. The 2013 International Society for Heart and Lung Transplantation Guidelines for mechanical circulatory support: executive summary. J Heart Lung Transplant. 2013;32(2):157–87.
Crawford TC, Okada DR, Magruder JT, Fraser C, Patel N, Houston BA, et al. A contemporary analysis of heart transplantation and bridge-to-transplant mechanical circulatory support outcomes in cardiac sarcoidosis. J Card Fail. 2018;24(6):384–91.
•• Jackson KC, Youmans QR, Wu T, Harap R, Anderson AS, Chicos A, et al. Heart transplantation outcomes in cardiac sarcoidosis. J Heart Lung Transplant. 2022;41(1):113–22. This retrospective study showed patients with CS have similar survival after heart transplantation and similar rates of graft failure and post-transplant malignancy as patients without CS.
Asleh R, Briasoulis A, Doulamis I, Alnsasra H, Tzani A, Alvarez P, et al. Outcomes after heart transplantation in patients with cardiac sarcoidosis. ESC Heart Fail. 2022;9(2):1167–74.
Yager JE, Hernandez AF, Steenbergen C, Persing B, Russell SD, Milano C, et al. Recurrence of cardiac sarcoidosis in a heart transplant recipient. J Heart Lung Transplant. 2005;24(11):1988–90.
Funding
This work was supported by a grant from the Ann Theodore Foundation to RK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Karen Flores Rosaio; Kyla Brezitski; Kelly Arps; Megan Milne; Jayanth Doss; and Ravi Karra declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Autoimmunity
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rosario, K.F., Brezitski, K., Arps, K. et al. Cardiac Sarcoidosis: Current Approaches to Diagnosis and Management. Curr Allergy Asthma Rep 22, 171–182 (2022). https://doi.org/10.1007/s11882-022-01046-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11882-022-01046-x