
Abstract
Epidemiologic studies highlight the increasing prevalence of vitamin D deficiency and insufficiency and its association with an increased risk of autoimmune diseases and poor respiratory function, including asthma. These and additional studies have raised interest in the immunomodulatory properties of vitamin D beyond its well-established role in calcium homeostasis and bone health. Vitamin D has been shown to influence the function of cells intrinsic to innate and adaptive immunity. This review discusses recent evidence that vitamin D promotes—both directly and indirectly—regulatory or suppressor T-cell populations with the capacity to inhibit inappropriate immune responses that cause disease, suggesting that this property may in part underpin the epidemiologic findings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vitamin D
The active form of vitamin D, 1α,25-dihydroxyvitamin D3 (1,25-[OH]2D3), is a secosteroid hormone that is mainly generated by a sunlight-catalyzed biosynthesis pathway that begins in the skin. UVB radiation (wavelengths, 270–300 nm) is absorbed by the epidermal and dermal cells and splits the B ring of 7-dehydrocholesterol, leading to the production of pre–vitamin D3. Pre–vitamin D3 is then rapidly converted to vitamin D3, which leaves the skin and enters the liver. In the liver, it is converted to 25-hydroxyvitamin D by cytochrome P450 enzymes (25-hydroxylases). 25-Hydroxyvitamin D is a circulating metabolite that is converted to the active form, 1,25-(OH)2D3, using the mitochondrial enzyme 25-hydroxyvitamin D-1α-hydroxylase [1]. The conversion of 25-hydroxyvitamin D to 1,25-(OH)2D3 was originally thought to occur mainly in kidney cells, but there is increasing evidence of extrarenal sources of 1,25-(OH)2D3 facilitated by cells including macrophages, epithelial cells, and dendritic cells (DCs), which may represent an important source of vitamin D for immunomodulatory actions in tissues [1, 2, 3•].
1,25-(OH)2D3 binds to the vitamin D receptor (VDR), a member of the superfamily of nuclear hormone receptors; this leads to a conformational change in the VDR that results in binding to the retinoic X receptor (RXR) and formation of a heterodimer. This heterodimer translocates to the nucleus, where it can bind to a vitamin D response element and promotes transcription of vitamin D–responsive genes. The VDR–RXR heterodimer can also bind to a “negative” vitamin D response element and prevent gene transcription, or it can bind to transcription factors present in the nucleus and prevent binding to a target gene promoter [1]. VDR is expressed by many cells of the immune system, including activated B and T cells, monocytes, and DCs. This, together with epidemiologic evidence discussed subsequently, has aroused considerable interest in the immunomodulatory properties of vitamin D, including its capacity to promote regulatory T-cell (Treg) populations.
Vitamin D status is commonly measured by assessing the serum concentration of 25-hydroxyvitamin D. Although outside the scope of this review, there is ongoing discussion on which levels define deficiency, insufficiency, and sufficiency. However, vitamin D deficiency (<50 nmol/L) has been historically linked to bone conditions such as rickets and osteomalacia and is also important in osteoporosis and fracture risk [4]. More recently, widespread reports have identified that vitamin D insufficiency (between 50 and 75 nmol/L) is at levels thought likely to impact on immune function and is on the increase within the developed world due to changes in lifestyle during the past few decades. For example, a recent study demonstrated that during the winter and spring months, 87% of the United Kingdom population is vitamin D insufficient [2, 5]. However, vitamin D insufficiency is relatively common, even in equatorial regions in which lower levels of vitamin D in children were associated with increased markers of asthma severity [6]. Several studies have reported associations of low vitamin D status with poor pulmonary function and an increased incidence of respiratory infections, asthma, and chronic obstructive pulmonary disease [6–8].
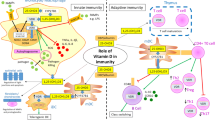
Vitamin D sufficiency is associated with a reduced risk of colorectal and other cancers, while insufficiency has been associated with a range of autoimmune disorders, including multiple sclerosis, type 1 diabetes mellitus, inflammatory bowel disease, and rheumatoid arthritis [8]. These epidemiologic data highlighting the link between vitamin D insufficiency and a range of immune-mediated disorders have emerged in parallel with experimental studies on the immunomodulatory properties of vitamin D, in particular its capacity to inhibit effector T-cell responses and to promote Tregs, which may, at least in part, begin to explain some of these associations. Figure 1 is a schematic of mechanisms by which vitamin D promotes peripheral tolerance while maintaining innate immune mechanisms that are important for the defense against infection.

Effects of vitamin D on cells of the innate and adaptive immune response likely to promote peripheral tolerance while maintaining innate immune mechanisms important for the defense against infection. Vitamin D promotes an antimicrobial environment via induction of antimicrobial peptides such as cathelicidin and β2-defensin. In conjunction with this, vitamin D renders dendritic cells (DCs) more immature via downregulation of costimulatory molecules HLA-DR, CD86, CD80, and the maturation marker CD83, and inhibition of the T-helper type 1 (Th1)-skewing cytokine interleukin (IL)-12. It also induces expression of inhibitory ligands such as immunoglobulin-like transcript 3 (ILT3) and programmed cell death 1 ligand 1 (PD-L1), and the anti-inflammatory cytokine IL-10. DCs are rendered tolerogenic and promote FoxP3+ and IL-10+ regulatory T cells (Tregs). Vitamin D also acts directly on T cells to promote FoxP3+ and IL-10+ Tregs, secretion of the immunomodulatory cytokines IL-10 and transforming growth factor (TGF)-β, and upregulation of the inhibitory molecule cytotoxic T-lymphocyte antigen (CTLA)-4. These Tregs subsequently inhibit innate and adaptive immune responses by downregulating proinflammatory cytokines, upregulating IL-10 and TGF-β, and via effects on a range of cell types, including antigen-presenting cells and T cells. aThere is a lack of consensus on the effects of vitamin D on Th2 and IgE allergic-type responses, but recent data may provide an explanation demonstrating significant but nonlinear effects of vitamin D on these parameters. IFN—interferon; TLR—Toll-like receptor
Regulatory T Cells
Several different Treg populations have been described with the capacity to inhibit a diverse array of immune responses and maintain immunologic tolerance in the periphery. The general belief is that Tregs exist in balance with effector T cells [9]. While effector T cells are essential for the elimination of many pathogens, they may also cause immune pathology through overexuberant or uncontrolled responses to pathogens, or inappropriate responses to self-antigens, commensal bacteria, and harmless environmental antigens such as allergens.
Although many cell types have the capacity to inhibit immune responses, most commonly via the production of immunosuppressive or anti-inflammatory mediators, considerable focus has been placed on T cells, particularly the CD4+ T-cell subset, in part due to their capacity for antigen specificity. Natural Tregs that are selected in the thymus are thought to play an important role in maintaining peripheral tolerance and inhibiting immune responses to self-antigens. Additional mechanisms for the peripheral induction of Tregs are likely to be important to maintain the Treg population as the thymus involutes with age, and to generate Tregs with a wider array of antigen specificities.
The concept that T cells can suppress as well as enhance immune responses has been recognized for a considerable time. However, the “modern era” of Tregs emerged from studies by Sakaguchi and others in the mid-1990s [9, 10], as they described “natural” Tregs produced in the thymus. These cells were characterized by the constitutively high expression of the CD25 antigen, the α-chain of the interleukin (IL)-2 growth factor receptor. CD25 is normally only transiently expressed at moderate levels on effector T cells upon activation. Further work identified—first in mice and subsequently confirmed in humans—that the lineage-specific transcription factor for these cells was forkhead box P3 (FoxP3) [11]. Nevertheless, identification of cell surface antigens that uniquely distinguish Tregs from T effector cells or differentiate between different Treg populations has proven elusive. “Naturally” occurring FoxP3+ Tregs are vitally important for self-tolerance and immune homeostasis, as best highlighted by cases of immune dysregulation polyendocrinopathy, enteropathy X-linked syndrome (IPEX), in which patients lack Tregs due to mutations in the gene FOXP3. IPEX is a fatal condition due to widespread autoimmune, allergic, and inflammatory disease from a very early age unless it is treated aggressively by bone marrow transplantation or profound immune suppression. A similar condition, scurfy, has been described in mice [11].
Adaptive Tregs are generated in the periphery via several different experimental protocols often associated with suboptimal activation of immune responses, such as via immature or alternatively activated antigen-presenting cell (APC) populations [12]. The capacity of effector T cells to deviate toward a Treg phenotype is also increasingly being described [13]. Adaptive or induced Tregs comprise both FoxP3+ and FoxP3- populations and synthesize anti-inflammatory mediators such as IL-10, transforming growth factor (TGF)-β, IL-35, and adenosine. The various Treg populations may additionally control immune responses through mechanisms including inhibitory cell surface ligands such as cytotoxic T-lymphocyte antigen (CTLA)-4 and programmed cell death 1 ligand 1 (PD-L1), cytotoxic mediators such as granzyme A and B, competition for growth factors, or via metabolic disruption involving CD39 and CD73 [14]. The range of inhibitory mechanisms used by Tregs has been the subject of recent excellent reviews [14].
Effects of Vitamin D on Effector T-cell Responses and Antigen-Presenting Cell Function
The magnitude and nature of the T-cell response is dependent on the context in which antigen is presented to T cells by dedicated APCs. DCs in particular play a central role in this process and are viewed as a pivotal link between innate and adaptive immunity. Innate pattern-recognition receptors expressed by DCs trigger their activation and are central for skewing the adaptive immune responses that are subsequently induced. Additional local environmental signals, such as inflammatory cytokines and in all likelihood 1,25-(OH)2D3, influence DC phenotype and activation and thus the nature of the T-cell response (eg, T-helper type 1 [Th1], Th2, Th17, Treg). Conversely, immature DCs with poor stimulatory function for the induction of effector T-cell responses often drive Treg responses.
Most of the direct immunomodulatory properties of 1,25-(OH)2D3 on DCs are proposed to occur in myeloid (mDCs) and not plasmacytoid DCs (pDCs). A study by Penna et al. [15] suggests that although both primary human blood–derived mDCs and pDCs express comparable levels of VDR and upregulate the primary response gene CYP24 upon culture with 1,25-(OH)2D3, only the tolerogenic properties of mDCs are modulated upon culture with 1,25-(OH)2D3. A recent study of gene profiling of vitamin D–treated DCs suggests that 1,25-(OH)2D3 upregulates several target genes that render the DCs more immature. These data identify complex patterns of gene expression via microarray analysis. Importantly, these data have been validated in part on primary human peripheral blood mDCs [16].
In functional studies, 1,25-(OH)2D3 treatment of human DCs in vitro results in reduced expression of the costimulatory molecules CD80 and CD86 and decreased expression of HLA-DR and the maturation marker CD83, all associated with an immature DC phenotype. 1,25(OH)2D3 also affects DC maturation from monocytes through inhibition of the production of IL-12 p70 and enhanced IL-10 secretion upon activation by CD40 ligation [17, 18]. 1,25-(OH)2D3 induces expression of immunoglobulin-like transcript 3 (ILT3) in vitro, an inhibitory receptor containing three cytoplasmic immunoreceptor tyrosine-based inhibitory receptor motifs, which when activated by the ligand result in an intracellular inhibitory signaling cascade [19]. The relevance of this in vitro observation is highlighted by studies in patients with psoriasis, in whom topical treatment of their lesions with 1,25-(OH)2D3 resulted in increased expression of ILT3 on DCs in conjunction with a reduction in the histology score of the lesions [19]. In vitro, 1,25-(OH)2D3-induced ILT3 appears to play a role in inhibiting T-cell responsiveness, as blockade of ILT3 on 1,25-(OH)2D3-treated DCs resulted in increased production of interferon (IFN)-γ [19]. In addition, upon activation, monocyte-derived DCs treated with 1,25-(OH)2D3 express enhanced levels of PD-L1, blockade of which significantly increased IFN-γ secretion in T-cell co-cultures [20].
Vitamin D inhibits effector T-cell responses via modulation of APC function and via direct inhibition of T-cell responses. The active form of vitamin D (1,25-[OH]2D3) decreases the production of the autocrine T-cell growth factor IL-2 via inhibition of the transcription factor known as nuclear factor of activated T cells, and this leads to a marked reduction in CD4+ T-cell proliferation [21]. 1,25-(OH)2D3 inhibits Th1 cytokine release with a large reduction in the production of IFN-γ from human peripheral blood CD3+CD4+ T cells [22]. 1,25-(OH)2D3 also hinders cytokine production by Th17 cells in murine autoimmune disease models, as well as in human CD4+ T cells in vitro [23, 24].
The effects of vitamin D on Th2 responses are less clear, with reports of both inhibition and enhancement. Early murine model data, including the use of a VDR knockout mouse, suggested that 1,25-(OH)2D3 enhanced the development of Th2 cells [25, 26]. A study using human peripheral blood mononuclear cells demonstrated that active vitamin D at a single very high dose enhanced Th2 cytokine production [27]. However, other studies in naive murine and human T cells reported that 1,25-(OH)2D3 inhibited the expression of IL-4 and other Th2 cytokines in vitro [28, 29]. It is notable that in animal models in which an increased Th2 response was observed [23, 25], this was accompanied by an increase in IL-10, symptomatic of a regulatory rather than an inflammatory Th2 phenotype [30]. Results of two recent human studies, one assessing serum and vitamin D status IgE [31•] and a second looking at a broad dose response of active vitamin D on human CD4+ T cells in culture [32], suggest a nonlinear effect of vitamin D on Th2 responses. These reports may help explain the earlier divergent data, suggesting overall that only abnormally low or high levels of vitamin D will adversely influence Th2 responses. Of additional interest here, Kreindler et al. [33] recently reported that in patients with allergic bronchopulmonary aspergillosis, heightened Th2 reactivity, assessed as serum IL-5 levels, correlated with low vitamin D status [33]. Similarly, ingestion of calcitriol (the active form of vitamin D, 1,25-[OH]2D3) by steroid-refractory asthma patients failed to increase Th2 cytokine gene expression in CD3+CD4+ T cells analyzed directly ex vivo but did increase IL-10 gene expression [32]. These data suggest, together with other studies discussed subsequently, that concomitant with the inhibition of an effector response is the induction of tolerance and Treg populations.
Vitamin D and Regulatory T Cells
A range of experimental approaches have been used to highlight the capacity of vitamin D to modulate FoxP3+ and IL-10+ Treg numbers and/or function. These range from in vitro and in vivo murine models, to patient-based studies demonstrating associations between vitamin D status and Treg numbers and/or function, and studies assessing Treg status before and after delivery of vitamin D to patients through supplementation with the parent compound or pharmacologic delivery of the active moiety. Together, these data have generated considerable interest in the therapeutic application of vitamin D to enhance Treg function from early life through to adulthood in a wide range of immune-mediated disorders.
Human In Vitro Studies
Inhibition of effector or inflammatory responses is frequently paralleled by the induction of a tolerogenic response and of Tregs. In this context, several studies have shown that pretreatment of human blood–derived mDCs, both monocyte-derived and primary mDCs, with 1,25-(OH)2D3 and then co-cultured with T cells not only inhibited T-effector cytokine production but also induced CD4+FoxP3+ cells with suppressive activity [19, 34•]. However, ILT3 expression induced by 1,25-(OH)2D3 on DCs in culture appeared to be dispensable for the capacity of 1,25-(OH)2D3-treated DCs to induce CD4+FoxP3+ Tregs, which is in contrast to its proposed role in modulation of the Th1 cytokine response [19].
1,25-(OH)2D3 may additionally act directly on T cells to promote a Treg phenotype. In bulk cultures of human CD4+CD25- T cells and putative naïve T cells, 1,25-(OH)2D3 was reported to increase the frequency of activation-induced FoxP3+ T cells, and this was dependent on the presence of IL-2 in culture. These FoxP3+ Tregs expressed high levels of CTLA-4, an inhibitory receptor. A significant reduction in the proinflammatory cytokines IFN-γ and IL-17 was also observed [35]. However, a comparable study using naïve murine T cells suggests that 1,25-(OH)2D3 inhibits both IL-17 and Treg differentiation in vitro [36].
A significant body of literature describes the capacity of retinoic acid to induce FoxP3+ Tregs, originally stemming from studies in murine gut [37]. Retinoic acid and vitamin D are members of the same nuclear hormone family and share a common signaling receptor, RXR [1]. It is presently unclear whether retinoic acid and vitamin D play complementary or redundant roles in Treg induction, or whether they play a comparable role in mice and humans. Furthermore, an emerging concept is the plasticity within the T-cell lineage and the capacity of previously activated effector T cells and differentiated T-cell lineages to convert to a regulatory phenotype [13]. Thus, it also will be of interest to determine, for example, whether vitamin D and retinoic acid target the same T-cell population (eg, naive vs previously activated T cells). Finally, differential effects of these two mediators on T-cell homing, referenced below, may indicate different roles in distinct mucosal compartments.
Vitamin D positively influences the IL-10–secreting Treg populations. Unger et al. [20] demonstrated that pretreatment with 1,25-(OH)2D3 upregulates expression of PD-L1, an inhibitory receptor, on monocyte-derived DCs and that these modified DCs were able, upon co-culture, to convert CD4+ T cells into IL-10–secreting Tregs capable of suppressing the proliferation of responder T cells [20]. In our own studies, activation of human and murine CD3+CD4+ T cells in vitro in the presence of a combination of the glucocorticosteroid dexamethasone (Dex) and 1,25-(OH)2D3 induced a sizeable population of IL-10–secreting Tregs. The regulatory capacity of these IL-10–secreting Tregs generated in vitro was assessed in vivo in a murine model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE). Transfer of the IL-10+ Tregs resulted in prevention of EAE in this model. The cells equally inhibited human Th1 and Th2 responses in vitro, and this was reversible through inhibition of IL-10 signaling in culture [38].
Subsequent studies demonstrated that human peripheral blood CD4+ T cells polyclonally activated in the presence of 1,25-(OH)2D3 alone also promoted IL-10–secreting Tregs. The microbial pattern-recognition receptor known as Toll-like receptor (TLR)9 was described as a biomarker of vitamin D–induced IL-10+ Tregs, and ligation of TLR9 with its agonist CpG oligonucleotide turned off IL-10 production, suggesting a control mechanism whereby Treg function may be abrogated [32]. This may be of relevance, for example, during infection, in which the capacity to transiently block inhibitory function would facilitate a more effective immune response for clearance of the pathogen. Elimination of the pathogen would lead to diminished TLR ligation, allowing restoration of the Treg response and thereby minimizing tissue damage [39]. Expression of microbial pattern-recognition receptors is more commonly associated with cells of the innate immune response. Schauber et al. [40] found that 1,25-(OH)2D3 acted upon keratinocytes to increase expression of the microbial pattern-recognition receptors TLR2 and CD14 and upregulate cathelicidin. The authors proposed that vitamin D acts in innate immunity also, enabling keratinocytes to recognize and respond to microbes and to protect wounds against infection. Thus, vitamin D deficiency may be important in the predisposition of skin from patients with atopic dermatitis to superinfection by Staphylococcus aureus [40].
The capacity of 1,25-(OH)2D3 to induce IL-10 and TLR9 on human CD4+ T cells was also demonstrated directly in patients. CD3+CD4+ T cells were analyzed directly ex vivo from steroid-refractory asthma patients before and after calcitriol treatment at standard formulary doses. Increased IL-10 and TLR9 gene expression was observed following calcitriol ingestion [32]. Earlier work on human cord blood determined that samples obtained during the summer months (hence higher levels of serum 25-hydroxyvitamin D) had a significantly higher level of IL-10 compared with samples obtained during winter months with lower serum vitamin D [41], further supporting the observation that vitamin D increases IL-10 in vivo.
These studies were shown to have clear application to human disease, specifically asthma. Earlier observations demonstrated that steroids induce IL-10 synthesis via human CD4+ T cells from healthy donors and steroid-sensitive asthma patients, but not in asthma patients who failed to give a good clinical response to steroids for improvement of their lung function—termed steroid insensitive or refractory—suggesting that the induction of IL-10 might contribute to the clinical efficacy of steroids in asthma [42]. The culture of peripheral blood T cells from steroid-refractory asthma patients with both Dex and active vitamin D completely restored the defective steroid-induced IL-10 response to levels observed in cultures of cells from steroid-sensitive asthmatics that were cultured with Dex alone. More strikingly, ingestion of oral 1,25-(OH)2D3 (calcitriol) by steroid-refractory asthmatics restored the capacity of the T cells to produce IL-10 in vitro in response to Dex. Mechanistic studies suggested that 1,25-(OH)2D3 influenced downregulation of the glucocorticoid receptor by its ligand [43]. These studies suggest that vitamin D may have therapeutic potential in severe asthma patients as a steroid-enhancing agent and are complemented by emerging studies showing an association between low vitamin D status and poor clinical responsiveness to glucocorticoids in asthma patients [6, 44]. If a steroid-enhancing role for vitamin D is proven, this is likely to be applicable to other chronic illness in which steroids represent a primary treatment, as also suggested by animal model data [23].
In conjunction with 1,25-(OH)2D3 effects on T cells, a recent article showed that 1,25-(OH)2D3 treatment of human B cells enhanced production of the immunomodulatory cytokine IL-10 [45]. 1,25-(OH)2D3 may therefore have an additional regulatory effect on B cells, potentially converting B cells into cells with a more regulatory role, although these concepts are still preliminary.
Animal Model Studies
Animal models have provided a wealth of data, largely in agreement with human laboratory studies, on the capacity of vitamin D to promote Tregs. Although subtle differences exist in the data from the varying models, in general, these demonstrate that irrespective of the route of administration, vitamin D mediates the expansion of Tregs and amelioration of immune-mediated diseases in vivo. In vitro and in vivo data provide evidence for promotion of tolerogenic DCs and enhanced IL-10, TGF-β, FoxP3, and CTLA-4 expression.
Early murine studies demonstrated that 1,25-(OH)2D3 alone or in combination with immunosuppressive drugs enhanced CD25+FoxP3+ Tregs in vivo. In a transplant model, a combination of oral mycophenolate mofetil (selective T- and B-cell inhibitor) and 1,25-(OH)2D3 prior to and during transplantation resulted in acceptance of and tolerance to a fully mismatched mouse islet allograft [46]. This tolerance was associated with decreased DC expression of CD40, CD80, CD86 and IL-12; reduced Th1 cytokine production; and an increased frequency of CD25+CD4+CD152+ Tregs, which could transfer transplant tolerance to a recipient mouse.
Vitamin D has beneficial effects in many animal models of autoimmune disease, including antiretinal autoimmunity [24], acute colitis [23], diabetes [47], arthritis [48], and EAE [34•, 36]. In a study of the progression of diabetes in nonobese diabetic mice, 1,25-(OH)2D3 alone inhibited Th1 infiltration and the progression of diabetes associated with an increased frequency of CD4+CD25+ Tregs in pancreatic lymph nodes [47]. In an EAE model, oral administration of active vitamin D prevented disease onset and CD4+ T cells in the central nervous system and decreased IL-17+ cells in the spleen and central nervous system, albeit with no change in IL-10 or IFN-γ and a slight decrease in FoxP3+ T cells in the spleen. In vitro, the authors described increased IL-10 production but inhibition of FoxP3 by vitamin D due to the inhibition of IL-2 required for FoxP3- Treg induction [36]. In an acute colitis model, intraperitoneal administration of calcitriol alone, or more effectively with Dex, reduced the severity of disease and local Th1 parameters. Locally increased IL-10, TGF-β, FoxP3, and CTLA-4 were all reported. The authors concluded that their data support a steroid-sparing clinical application for calcitriol derivatives in inflammatory bowel disease [23].
Two independent groups suggest that topical administration of 1,25-(OH)2D3 to the skin of mice resulted in the in vivo expansion of antigen-specific CD25+CD4+FoxP3+ Tregs in the skin-draining lymph node [49, 50]. These Tregs demonstrated an enhanced capacity to suppress immune responses in vitro and in vivo, and these data are clearly relevant to the topical use of active vitamin D in psoriasis [19].
In contrast, some early studies suggested that vitamin D has deleterious effects in allergic airway disease. VDR-deficient mice failed to develop experimental allergic asthma, leading the authors to suggest a role for vitamin D in driving Th2 inflammation in the airways [26]. Nevertheless, considerable interest remains in the therapeutic application in asthma, and examples of beneficial effects exist [51]. In a murine model of allergic airway disease, 1,25-(OH)2D3 potentiated the effects of a suboptimal allergen (ovalbumin) immunotherapy regimen to ameliorate airway hyperresponsiveness and to reduce eosinophilia, Th2 responses, and IgE (in serum) via induction of the regulatory cytokines IL-10 and TGF-β [52]. When human CD4+ T cells obtained from allergic bronchopulmonary aspergillosis (Th2-mediated disease) patients were cultured with the addition of 1,25-(OH)2D3, reduced DC expression of OX40L and increased TGF-β expression by DCs and Tregs were reported, together with inhibition of Th2 responses [33].
1,25-(OH)2D3 not only influences the generation of tolerogenic immune responses but also expression of chemokines and their receptors that are likely to determine T-cell homing. For example, 1,25-(OH)2D3 signaled T cells to express CC chemokine receptor (CCR)10, which enabled them to migrate to the skin-specific chemokine CCL27 secreted by keratinocytes of the epidermis but suppressed the gut-homing receptors α4β7 and CCR9, a property more commonly associated with retinoic acid [53]. 1,25-(OH)2D3 inhibits the expression of CCL17 and CCL20 but induces expression of CCL22 in mDCs that are suggested to play a key role in the recruitment of Tregs [15]. Clearly defining the effects of vitamin D on T-cell homing is an important component in our understanding of its importance in the regulation of immunity.
Therapeutic Application of Vitamin D in Humans: Evidence for Immunomodulatory Actions
Epidemiologic studies highlighting associations with vitamin D status and various immune disorders have heightened interest in addressing further correlations with immunoregulatory cytokines and Treg activity in patients. For example, recent evidence shows that in multiple sclerosis patients, low serum 25-hydroxyvitamin D status (but not serum 1,25-dihydroxyvitamin D3) was associated with reduced suppressive capacity of CD25+CD4+CD127lo Tregs [54]. Similar associations have been shown for IL-10 [41]. In humans, vitamin D supplementation and pharmacologic treatment with active vitamin D have been shown to increase serum- or T-cell–associated TGF-β and IL-10, respectively [32, 55].
Conclusions
The active form of vitamin D (1,25-[OH]2D3) influences innate and adaptive immunity. It acts on APCs and T cells to promote peripheral tolerance via inhibition of inflammatory responses and induction of Tregs. Growing interest exists in the physiologic role of vitamin D as an essential mediator in maintaining a healthy and functional immune system. However, many questions remain regarding its role compared with other related compounds such as retinoic acid. Therapeutic evaluation of vitamin D supplementation and active vitamin D, including analogues with reduced calcemic effects, is ongoing. Studies of this nature will be crucial to furthering our understanding of both the physiologic role of vitamin D in the maintenance of health and its therapeutic evaluation, including steroid-enhancing activity, for a range of immune and inflammatory disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Mora JR, Iwata M, von Andrian UH: Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol 2008:685–698.
Adams JS, Hewison M: Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat Clin Pract Endocrinol Metab 2008, 4:80–90.
• Hewison M: Vitamin D and the intracrinology of innate immunity. Mol Cell Endocrinol 2010, 321:103–111. This is an excellent overview highlighting the capacity for local synthesis of vitamin D in tissues and the effects on innate immunity.
Nassef M, Temparano J, Frieri M et al: Should Fracture Risk Influence Our Decision Making in Asthma Care? . Ann Allergy Asthma Immunol 2010, In press.
Hypponen E, Power C: Hypovitaminosis D in British adults at age 45 y: nationwide cohort study of dietary and lifestyle predictors. Am J Clin Nutr 2007, 85:860–868.
Brehm J M, Celedon J C, Soto-Quiros M E et al: Serum vitamin D levels and markers of severity of childhood asthma in Costa Rica. Am J Respir Crit Care Med 2009, 179:765–771.
Black P N, Scragg R: Relationship between serum 25-hydroxyvitamin D and pulmonary function in the third national health and nutrition examination survey. Chest 2005, 128:3792–3798.
Holick M: Vitamin D deficiency. The New England Journal of Medicine 2007, 357:266–281.
Sakaguchi S, Miyara M, Costantino CM, Hafler DA: FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 2010, 10:490–500.
Sakaguchi S, Sakaguchi N, Asano M et al: Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995, 155:1151–1164.
Torgerson TR, Ochs HD: Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol 2007, 120:744-750; quiz 751–742.
Hawrylowicz CM, O’Garra A: Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat Rev Immunol 2005, 5:271–283.
Murphy KM, Stockinger B: Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol 2010, 11:674–680.
Vignali DA, Collison LW, Workman CJ: How regulatory T cells work. Nat Rev Immunol 2008, 8:523–532.
Penna G, S Amuchastegui, N Giarratana, K C Daniel, M Vulcano, S Sozzani, Adorini L: 1,25-Dihydroxyvitamin D3 selectively modulates tolerogenic properties in myeloid but not plasmacytoid dendritic cells. The Journal of Immunology 2007, 178:145–153.
Szeles L, G Keresztes, D Torocsik, Z Balajthy, L Krenacs, S Poliska, A Steinmeyer, U Zuegel, M Pruenster, A Rot, et al.: 1,25-dihydroxyvitamin D3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype. The Journal of Immunology 2009, 182:2074–2083.
Penna G, Adorini L: 1 Alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol 2000, 164:2405–2411.
Canning MO, Grotenhuis K, de Wit H et al: 1-alpha,25-Dihydroxyvitamin D3 (1,25(OH)(2)D(3)) hampers the maturation of fully active immature dendritic cells from monocytes. Eur J Endocrinol 2001, 145:351–357.
Penna G, Roncari A, Amuchastegui S et al: Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood 2005, 106:3490–3497.
Unger WW, Laban S, Kleijwegt FS et al: Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol 2009, 39:3147–3159.
Alroy I, T L Towers, Freedman LP: Transcriptional repression of the interleukin-2 gene by vitamin D3: direct inhibition of NFATp/AP-1 complex formation by a nuclear hormone receptor. Molecular and Cellular Biology 1995, 15:5789–5799.
Reichel H, Koeffler HP, Tobler A, Norman AW: 1 alpha,25-Dihydroxyvitamin D3 inhibits gamma-interferon synthesis by normal human peripheral blood lymphocytes. Proc Natl Acad Sci U S A 1987, 84:3385–3389.
Daniel C, Sartory NA, Zahn N et al: Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J Pharmacol Exp Ther 2008, 324:23–33.
Tang J, Zhou R, Luger D et al: Calcitriol suppresses antiretinal autoimmunity through inhibitory effects on the Th17 effector response. J Immunol 2009, 182:4624–4632.
Boonstra A, Barrat FJ, Crain C et al: 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol 2001, 167:4974–4980.
Wittke A, Weaver V, Mahon BD et al: Vitamin D receptor-deficient mice fail to develop experimental allergic asthma. J Immunol 2004, 173:3432–3436.
Jirapongsananuruk O, Melamed I, Leung DY: Additive immunosuppressive effects of 1,25-dihydroxyvitamin D3 and corticosteroids on TH1, but not TH2, responses. J Allergy Clin Immunol 2000, 106:981–985.
Pichler J, Gerstmayr M, Szepfalusi Z et al: 1 alpha,25(OH)2D3 inhibits not only Th1 but also Th2 differentiation in human cord blood T cells. Pediatr Res 2002, 52:12–18.
Staeva-Vieira TP, Freedman LP: 1,25-dihydroxyvitamin D3 inhibits IFN-gamma and IL-4 levels during in vitro polarization of primary murine CD4+ T cells. J Immunol 2002, 168:1181–1189.
Ito T, Wang YH, Duramad O et al: TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med 2005, 202:1213–1223.
• Hypponen E, Berry DJ, Wjst M, Power C: Serum 25-hydroxyvitamin D and IgE - a significant but nonlinear relationship. Allergy 2009, 64:613–620. This study of patients, together with the article by Urry et al. [32], begins to make sense of the confusion in the scientific literature regarding the contradictory reports of vitamin D on Th2 allergic-type responses.
Urry Z, E Xystrakis, D F Richards et al: Ligation of TLR9 induced on human IL-10-secreting Tregs by 1α,25-dihyrdoxyvitamin D3 abrogates regulatory function. The journal of Clinical Investigation 2009, 119:387–398.
Kreindler JL, Steele C, Nguyen N et al.: Vitamin D3 attenuates Th2 responses to Aspergillus fumigatus mounted by CD4+ T cells from cystic fibrosis patients with allergic bronchopulmonary aspergillosis. J Clin Invest 2010.
• Adorini L, Penna G: Induction of tolerogenic dendritic cells by vitamin D receptor agonists. Handb Exp Pharmacol 2009:251–273. This is a review on the capacity of vitamin D to induce tolerogenic DCs with Treg responses from a group that has contributed extensively in this area.
Jeffery LE, Burke F, Mura M et al: 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol 2009, 183:5458–5467.
Chang JH, Cha HR, Lee DS et al: 1,25-Dihydroxyvitamin D3 inhibits the differentiation and migration of T(H)17 cells to protect against experimental autoimmune encephalomyelitis. PLoS One 2010, 5:e12925.
Mucida D, Park Y, Cheroutre H: From the diet to the nucleus: vitamin A and TGF-beta join efforts at the mucosal interface of the intestine. Semin Immunol 2009, 21:14–21.
Barrat F J, Cua D, Boonstra A et al: In vitro generation of interleukin 10-producing regulatory CD4+ T cells is induced by immunosuppressive drugs and inhibited by T helper 1 (Th1)- and Th2-induced cytokines. Journal of Experimental Medicine 2002, 195:603–616.
Sutmuller RP, Morgan ME, Netea MG et al: Toll-like receptors on regulatory T cells: expanding immune regulation. Trends Immunol 2006, 27:387–393.
Schauber J, Dorschner RA, Coda AB et al.: Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest 2007, 117:803–811.
Zittermann A, Dembinski J, Stehle P: Low vitamin D status is associated with low cord blood levels of the immunosuppressive cytokine interleukin-10. Pediatr Allergy Immunol 2004, 15:242–246.
Hawrylowicz C, Richards D, Loke T-K et al: A defect in corticosteroid-induced IL-10 production in T lymphocytes from corticosteroid-resistant asthmatic patients. The Journal of Allergy and Clinical Immunology 2002, 109:369–370.
Xystrakis E, Kusumakar S, Boswell S et al: Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. J Clin Invest 2006, 116:146–155.
Searing DA, Zhang Y, Murphy JR et al: Decreased serum vitamin D levels in children with asthma are associated with increased corticosteroid use. J Allergy Clin Immunol 2010, 125:995–1000.
Heine G, Niesner U, Chang HD et al: 1,25-dihydroxyvitamin D(3) promotes IL-10 production in human B cells. Eur J Immunol 2008, 38:2210–2218.
Gregori S, M Casorati, S Amuchastegui et al: Regulatory T cells induced by 1α,25-Dihydroxyvitamin D3 and mycophenolate mofetil treatment mediate transplantation tolerance. The Journal of Immunology 2001, 167:1945–1953.
Gregori S, N Giarratana, S Smiroldo et al: A 1α,25-dihydroxyvitamin D3 analog enhances regulatory T-cells and arrests autoimmune diabetets in NOD mice. Diabetes 2002, 51:1367–1374.
Cantorna MT, Hayes CE, DeLuca HF: 1,25-Dihydroxycholecalciferol inhibits the progression of arthritis in murine models of human arthritis. J Nutr 1998, 128:68–72.
Gorman S, Kuritzky LA, Judge MA et al: Topically applied 1,25-dihydroxyvitamin D3 enhances the suppressive activity of CD4+CD25+ cells in the draining lymph nodes. J Immunol 2007, 179:6273–6283.
Ghoreishi M, Bach P, Obst J et al: Expansion of antigen-specific regulatory T cells with the topical vitamin d analog calcipotriol. J Immunol 2009, 182:6071–6078.
Lloyd CM, Hawrylowicz CM: Regulatory T cells in asthma. Immunity 2009, 31:438–449.
Taher YA, van Esch BC, Hofman GA et al: 1alpha,25-dihydroxyvitamin D3 potentiates the beneficial effects of allergen immunotherapy in a mouse model of allergic asthma: role for IL-10 and TGF-beta. J Immunol 2008, 180:5211–5221.
Sigmundsdottir H, Pan J, Debes G F et al: DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nature Immunology 2007, 8:285–293.
Smolders J, Thewissen M, Peelen E et al: Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. PLoS One 2009, 4:e6635.
Mahon BD, Wittke A, Weaver V, Cantorna MT: The targets of vitamin D depend on the differentiation and activation status of CD4 positive T cells. J Cell Biochem 2003, 89:922–932.
Acknowledgments
Miss Chambers is a recipient of a Medical Research Council British Thoracic Society Capacity Building PhD studentship. The authors have received financial support from Asthma UK and the Department of Health via the National Institute for Health Research comprehensive Biomedical Research Centre award to Guy’s and St. Thomas’ National Health Service Foundation Trust in partnership with King’s College London and King’s College Hospital National Health Service Foundation Trust.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chambers, E.S., Hawrylowicz, C.M. The Impact of Vitamin D on Regulatory T Cells. Curr Allergy Asthma Rep 11, 29–36 (2011). https://doi.org/10.1007/s11882-010-0161-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11882-010-0161-8