
Abstract
It is well-known that autoimmunity is significantly more prevalent in females. Growing evidence indicates that genes located on the X chromosome may play a role in autoimmunity and immune dysregulation, as also indicated by the frequent association of autoimmune phenomena in patients with X-linked primary immune deficiencies (PIDs). Hence, this group of genetic disorders is of particular interest to study PID-causing genes in the setting of more complex autoimmune disorders. This review focuses on the mechanisms leading to the autoimmune phenomena that are associated with the different X-linked PIDs, and on the intriguing interplay between immune dysregulation and immune deficiency in this unique setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immune deficiencies, defined by abnormal or deficient immune development and/or function, are often associated with immune dysregulation and self-reactivity. Indeed, autoimmunity and immune deficiency are no longer viewed as two opposite extremes of immune function, but rather as intertwined phenomena that reflect inadequate immune function. Considerable research is focused on unraveling the mechanisms underlying this association [1, 2]. One of the earliest observations leading to this concept was the high prevalence of autoimmune diseases in females alongside the relative abundance of X-linked primary immune deficiencies (PIDs) [3•].
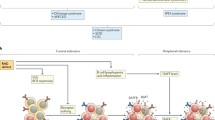
Currently, 11 X-linked genetic defects are known to result in PID (Fig. 1), and the clinical phenotype of most of them includes significant autoimmune manifestations (Table 1) [4••]. In an attempt to shed light on the intriguing association of immune deficiency and autoimmunity, this review focuses on the X-linked PIDs that are more frequently associated with autoimmune phenomena.

Primary immunodeficiency genes and their arrangement on the X chromosome. CGD—chronic granulomatous disease; EDA-ID—hypohidrotic ectodermal dysplasia with immunodeficiency; IKKg— IκB kinase-γ; IPEX—immune dysregulation, polyendocrinopathy, and enteropathy, X-linked syndrome; NEMO—nuclear factor-κB essential modulator; SCIDX1—X-linked severe combined immunodeficiency; WAS—Wiskott–Aldrich syndrome; XLA—X–linked agammaglobulinemia; XLP—X–linked lymphoproliferative disease
Immune Dysregulation, Polyendocrinopathy, and Enteropathy, X-Linked Syndrome
Immune dysregulation, polyendocrinopathy, and enteropathy, X-linked (IPEX) syndrome is characterized by early onset and severe immune dysregulation resulting in multiorgan damage. Patients with IPEX usually present within the first few weeks or months of life with severe enteropathy, dermatitis, and insulin-dependent diabetes mellitus [1, 2, 5]. In addition, some patients develop thrombocytopenia, hemolytic anemia, thyroiditis, and nephropathy. If untreated, patients with typical and severe clinical manifestations tend to die within the first few years of life.
A similar phenotype had been described in the naturally occurring “scurfy” mouse, with skin abnormalities, progressive enterocolitis with malabsorption, lymphadenopathy, and hepatosplenomegaly, as well as several hematologic abnormalities and inflammatory infiltrates in endocrine glands that led to death at 21–28 days of life [6].
IPEX and the scurfy mouse represent the archetype of monogenic disorders of immune overactivation and remain a key model for the study of regulatory T cells (Tregs) as well as of mechanisms of immune dysregulation in general.
Thymus-derived natural Tregs (nTregs) play a critical role in peripheral immune tolerance by suppressing self-reactive lymphocytes. Development of nTregs is controlled by the transcription factor forkhead box P3 (FoxP3), whose gene maps at Xq28. The clinical phenotype of IPEX and the “scurfy” mouse model had been known for many years, but it was not until the year 2000 that the molecular basis of IPEX, due to FOXP3 mutations, in humans was identified [7], followed shortly thereafter by confirmation of the Foxp3 mutation also in scurfy mice [8]. Further studies in scurfy mice showed that lack of Foxp3 expression abrogates development of thymus-derived CD4+CD25+ nTregs [9]. These observations were also confirmed in patients with IPEX [5]. Furthermore, it was shown that constitutive activation of CD4+ T lymphocytes plays a significant role in the pathogenesis of the autoimmune damage [6].
These discoveries paved the way for a better understanding of the central role of nTregs in mediating self-tolerance by suppressing activation of autoreactive T lymphocytes in the periphery [9, 10] and led to a better understanding of the pathophysiology of nTreg-associated autoimmunity.
Typically, a variety of autoantibodies directed against insulin, glutamic acid decarboxylase, enterocytes, and tissue-specific antigens are detected in patients with IPEX, but cell-mediated mechanisms also contribute to the severe tissue damage [1, 5].
Laboratory signs of immune dysregulation in patients with IPEX also include increased serum concentrations of IgE, whose levels often correlate with the severity of the clinical phenotype. This increased production of IgE and the associated eosinophilia may reflect an excess of T-helper type 2 (Th2) cytokines (interleukin [IL]-4, IL-5), and reduced secretion of the Th1 cytokine interferon (IFN)–γ also has been described [7].
The immune deficiency of IPEX is marked by increased occurrence of a wide range of infections (seen in up to 50% of the patients), and that may also include life-threatening episodes such as sepsis and meningitis [2, 5]. In spite of this, the FoxP3 defect seemingly does not impair immune defense mechanisms per se. Most patients can mount normal and protective specific antibody responses to immunization and maintain normal complement levels and neutrophil function [5]. They also show normal numbers and distribution of the major lymphocyte subsets, with preserved ability to respond to mitogens and antigens in vitro [2, 5]. This has led us to hypothesize that infections in patients with IPEX may be secondary to immune-mediated damage of mucosal tissue and barrier compromise and/or to use of immunosuppressive drugs that are required to control the severe autoimmune manifestations of the disease.
The prognosis for untreated patients is poor, and mortality is common within the first years of life. Although immune suppression may offer some benefit, hematopoietic cell transplantation remains the only curative treatment [5, 11].
Hypomorphic FOXP3 mutations that only partially hinder FoxP3 expression and/or function have been shown to result in a milder, delayed-onset phenotype that may include asthma, food allergies, and autoimmune hepatitis [2, 5, 12]. Furthermore, several patients have been described with IPEX-like features, which do not carry mutations in the FOXP3 gene. In few of them, biallelic mutations of the IL2RA (CD25) gene have been identified [13]. CD25 encodes the α chain of the IL-2 receptor, and IL-2 is important in sustaining development and function of nTregs [10]. However, most patients with IPEX-like features and no FOXP3 mutations remain genetically undefined.
Wiskott-Aldrich Syndrome
Wiskott-Aldrich syndrome (WAS) is caused by mutations in the WAS gene encoding for the WAS protein (WASp). WAS has an estimated frequency of 4 cases per 1 million births in males. The main characteristics of this rare X-linked PID are thrombocytopenia with small platelets and associated bruising and bleeding tendency, eczema, and susceptibility to infections, in particular due to encapsulated bacteria and herpes viruses [14, 15]. Patients are also at high risk for autoimmunity and malignancies.
However, significant variability of the clinical phenotype has been reported. Some patients present with a milder variant of the disease and a more favorable prognosis, known as X-linked thrombocytopenia (XLT) [16]. XLT is usually associated with hypomorphic mutations of the WAS gene that allow for residual protein expression and function, while loss of expression or function is usually associated with the complete WAS phenotype [14, 16]. Whereas WAS and XLT represent examples of loss-of-function mutations of the WAS gene, a distinct phenotype characterized by severe neutropenia and increased risk of myelodysplasia has been associated with gain-of-function mutations that result in constitutively activated forms of WASp expression [17].
WASp is expressed in all nonerythroid hematopoietic cells and functions in coupling cellular activation to actin polymerization and cytoskeleton remodeling [14, 15].
Both adaptive and innate immune responses, including Treg function, are affected by WAS mutations. In particular, WASp plays a major role in cell migration by facilitating formation of podosomes. Accordingly, chemokine-induced migration and trafficking of WASp-deficient monocytes, dendritic cells, neutrophils, as well as T and B lymphocytes is impaired [14].
The T-cell compartment is abnormal in WAS patients as a result of accelerated T-cell death that favors progressive T-cell lymphopenia. Reduced T-cell in vitro proliferation and IL-2 secretion upon T-cell receptor cross-linking also have been demonstrated [18]. Serum IgA and IgE levels tend to be above normal values, but IgM levels are usually low. Antibody responses to T-dependent and T-independent antigens are reduced [14]. Defective cytolytic natural killer (NK) cell function is seen and results from a markedly reduced accumulation of F-actin at the immunologic synapse and defective immunologic synapse formation [15].
Autoimmune manifestations are very common in WAS. Up to 70% of patients will develop one or more autoimmune disorders, including autoimmune hemolytic anemia, vasculitis, renal disease, Henoch-Schönlein–like purpura, and inflammatory bowel diseases. Less frequent are neutropenia, dermatomyositis, angioedema, and uveitis [14, 15]. Autoimmunity is more common in severely affected WASp-negative patients but also may be observed in patients with residual WASp expression [15, 16].
Various mechanisms may account for the increased occurrence of autoimmune manifestations in WAS. In particular, Treg function was shown to be reduced in both WASp-deficient humans and mice [19]. WASp-deficient Tregs express decreased levels of activation markers, lack several tissue-homing markers, and produce less of the immunosuppressive cytokine IL-10. Moreover, inability to clear pathogens, impaired phagocytosis, defective leukocyte trafficking, decreased production of IL-2 (which is important for Treg function), and increased production of autoantibodies owing to B-cell–intrinsic dysfunction all may contribute to autoimmunity [15].
WAS patients are also prone to malignancies (especially lymphomas and leukemias), with a 10-fold increase in the age-specific mortality rate for cancer compared with the general population. The risk of malignancies is even higher in WAS patients with autoimmune manifestations [14, 15].
X-Linked Lymphoproliferative Disease
X-linked lymphoproliferative disease (XLP) is characterized by extreme susceptibility to Epstein-Barr virus (EBV) infections. Patients remain asymptomatic until primary EBV infection, which results in an unrestrained immune response with polyclonal expansion of different lineages [20, 21]. Occasionally, other infectious agents such as cytomegalovirus, measles virus, and other pathogens, also may precipitate the XLP phenotype [21]. Various tissues are infiltrated by activated lymphocytes that produce an array of inflammatory mediators and cause substantial tissue damage.
XLP is a very severe disease [20, 21]. Patients may present with fulminant infectious mononucleosis, rapid liver and spleen enlargement, and liver necrosis. Alternatively, hemophagocytosis may lead to bone marrow failure. There is a high risk of development of lymphoma (especially large B-cell lymphoma). Mortality is extremely high, approaching 100% by age 20. A minority of patients may survive and develop progressive dysgammaglobulinemia, with reduced levels of IgG and increased serum concentrations of IgM.
To date, two distinct genetic defects have been found to be associated with XLP. The SH2 domain protein 1A (SH2D1A) that encodes for the signaling lymphocyte activation molecule (SLAM)-associated protein (SAP) is mutated in about 80% of XLP patients, while the X-linked inhibitor of apoptosis (XIAP) gene that is adjacent to SH2D1A at Xq25 is mutated in the remaining 20% [20–22].
SAP is an adaptor protein involved in intracellular signaling through receptors of the SLAM family that includes SLAM (CD150, the receptor for measles virus), 2B4 (CD244), NTB-A, and Ly9 (CD229). SAP is selectively expressed by T, NK, and NKT lymphocytes [20, 21]. SAP deficiency results in functional abnormalities in multiple lymphoid lineages. These include reduced CD8+ and NK lymphocytes’ cytotoxicity, altered follicular CD4+ T lymphocytes’ function, impaired NKT cell development, defective class-switched memory B-cell generation, and abnormal antibody production [20, 21].
Sap-deficient mice show vigorous proliferation and activation of CD8+ T cells upon infection with lymphocytic choriomeningitis virus or with γ-herpesvirus-68, with a dramatic skewing toward production of Th1 cytokines that leads to sustained inflammation and tissue damage [20, 23].
Intriguingly, XIAP-deficient patients do not present with most of the T- and NK-cell abnormalities described in SAP-deficient patients. They do, however, present with a lack of NKT lymphocytes [20, 22]. Furthermore, XIAP-deficient peripheral blood lymphocytes and immortalized B cells have increased sensitivity to apoptotic stimuli in vitro [22].
Although the mechanisms leading to the XLP phenotype in XIAP-deficient patients are not yet clear, it is possible that the NKT-cell defect, which is shared by SAP-deficient and XIAP-deficient patients, plays a key role in the development of susceptibility to EBV-induced lymphoproliferation, immune dysregulation, and hemophagocytic syndrome seen in these patients.
X-Linked Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is the result of deficient oxidative burst and impaired generation of reactive oxygen species by the NADPH oxidase complex due to mutations in any of the genes encoding for its different subunits. The X-linked form (X-CGD) results from mutations in the CYBB gene coding for NOX2 (gp91phox), while the remainder of the cases are caused by mutations in the autosomal genes that encode for other NADPH oxidase subunits [24]. Patients present with severe bacterial and fungal infections, usually within the first years of life. Other manifestations include colitis and multiple noninfectious granulomata, in some cases leading to secondary manifestations such as gastric outlet obstruction [24]. Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia spp, and Aspergillus spp are the most common pathogens [25]. Although recent advances and modern antibacterial and antifungal agents have significantly improved the prognosis of these patients, the disease carries significant morbidity and mortality, and hematopoietic cell transplantation remains an important curative option [26]. Several groups are investigating gene therapy approaches for X-CGD; these studies hold significant promise for the future [26, 27].
In recent years, longer follow-up of CGD patients yielded evidence for significant autoimmunity associated with CGD. CGD patients are prone to autoimmune diseases such as sarcoidosis, Crohn’s disease, and rheumatoid arthritis, as well as discoid lupus, systemic lupus erythematosus, idiopathic thrombocytopenic purpura, and Behcet’s syndrome [28–30]. Interestingly, female carriers of X-CGD are also prone to autoimmune manifestations such as lupus-like cutaneous manifestations, oral ulcers, Raynaud’s phenomenon, and a variety of joint symptoms. It is possible that these autoimmune manifestations in female carriers of X-CGD reflect the presence of mutant cells in the periphery due to random X chromosome inactivation [28, 30]. Similarly, murine models of X-CGD have been shown to be susceptible to noninfectious inflammatory processes in various organs, including joints, skin, digestive tract, brain, eyes, and lungs [30].
CGD patients also present with cellular abnormalities in the B-cell compartments, which include increased proportion of CD5+ B cells and a significantly decreased number of CD27+ B cells [31]. Activated dendritic cells from NADPH oxidase–deficient mice were shown to secrete higher levels of tumor necrosis factor (TNF)-α, IL-1β, IL-6, and transforming growth factor-β in vitro and to enhance the secretion of IL-17 and IFN-γ from responding T cells in vivo. These mice also show predisposition for T-cell–mediated inflammatory conditions [32]. Apoptosis processes were shown to be aberrant in neutrophils of CGD patients as well as in murine models of X-CGD. This was also shown to be associated with autoantibody production [33]. Controversy as to which of the phagosomal bactericidal mechanisms is more affected by the NADPH oxidase deficiency still exists, and the abnormal production of active superoxide metabolite, defective flux of potassium ions, and abnormal activity of neutrophil elastase and cathepsin G, among others, was suggested [25]. Regardless, the inability to efficiently clear phagocytized pathogens results in diversion of CGD cells from bactericidal function to a hyperinflammatory state, with increased production of IL-6 and TNF-α
CD40 Ligand Deficiency
CD40 ligand (CD40L) deficiency is associated with normal to increased serum IgM levels with very low levels of the other immunoglobulin subtypes [34]. It is part of a genetically heterogeneous spectrum of disorders of immunoglobulin class switching that may reflect B-cell–intrinsic defects or impaired Th cell activity [35]. CD40L deficiency is inherited as an X-linked trait, and the responsible gene (CD40LG) maps at Xq25 [36].
CD40L is a member of the TNF family. It is mostly expressed on activated CD4+ T cells. This allows these T cells to interact with CD40-expressing cells, mainly B cells, dendritic cells, and macrophages. Engagement of CD40 on the surface of B cells results in activation of the downstream signaling pathway that includes TNF receptor-associated factor 2 (TRAF2), TRAF3, and TRAF6 and leads to activation and nuclear translocation of nuclear factor-κB (NF-κB). As a result, expression of NF-κB-responsive genes such as AICDA and UNG is induced [2, 36]. The expression of these molecules is critical for immunoglobulin class switching. Expression of the costimulatory proteins CD80/CD86 on the surface of B lymphocytes and dendritic cells is also dependent on CD40L–CD40 interaction [37].
Thus, CD40L-deficient patients produce only IgM antibodies in response to T-dependent antigens and cannot generate memory B cells. They also have low numbers of CD45R0+ primed T cells as well as abnormal proliferation to antigens in vitro despite normal number and distribution of CD4+ and CD8+ subsets [35, 37]. Lymph nodes of CD40L-deficient patients contain primary follicles but lack germinal centers [36]. CD40–CD40L interaction was shown to be important in T-cell selection and maturation in the thymus, where CD40 is expressed by resident dendritic and B cells as well as on medullary and cortical thymic epithelial cells [34]. Furthermore, a reduced number of Aire-expressing (a transcription factor that plays a major role in deletion of autoreactive T cells) medullary thymic epithelial cells has been observed in CD40−/−mice [38]. CD40L–CD40 interaction was also shown to play a critical role in the production of Tregs [39]. Finally, peripheral B-cell tolerance is also affected in CD40L-deficient patients, in whom mature naive B cells were shown to express a high proportion of autoreactive antibodies [40]. Taken together, these observations suggest that CD40L–CD40 interaction is essential not only for normal immune function but also for maintaining normal central and peripheral tolerance. This may explain the fact that the clinical manifestation of CD40L deficiency resulting from these defects includes susceptibility to infections as well as various autoimmune manifestations. Patients present early in life with recurrent opportunistic infections and a tendency for developing gastrointestinal and biliary tract neoplasms [36]. Pneumocystis jiroveci and Cryptosporidium spp are among the typical infections [34, 36]. Autoimmune manifestations are frequent and involve joints, bowel, liver, skin, endocrine, hematopoietic, and vascular systems [35]. Antinuclear, anti-Ro, anti-RNP, anti-smooth muscle, anticardiolipin, antiplatelet, thyroid, and various other autoantibodies often can be detected.
Nuclear Factor-κB Essential Modulator/IKKγ Deficiency
Hypohidrotic ectodermal dysplasia (EDA) was recently found to be associated with immunodeficiency in a subset of patients with mutations of the IKBKG gene (EDA-ID) [41, 42]. About 20% to 25% of these patients also present with a hyper-IgM phenotype; hence, EDA-ID is also considered a part of the HIGM group (HIGM6). However, most patients present with variable levels of immunodeficiency without elevated IgM levels [43]. IKBKG encodes for the NF-κB essential modulator (NEMO)/IKKγ component of the IκB kinase (IKK) complex that plays a crucial role in the regulation of the NF-κB signaling pathway.
Whereas hypomorphic mutations of the IKBKG gene in males result in the EDA-ID phenotype, complete lack of IKBKG expression is lethal. Interestingly, females who are heterozygous for IKBKG null mutations present with incontinentia pigmenti [42, 43].
NF-κB is a key transcription factor responsible for the expression of many immune- and non-immune-related genes. The pathway leading to its activation is complex and may be triggered by various signal transduction signals initiated by different receptors, including Toll-like receptors (TLRs), members of the TNF receptor family (eg, CD40), and the B-cell receptor. Under resting conditions, NF-κB translocation to the nucleolus is prevented by IκB. Activatory signals that allow IKK-mediated phosphorylation of IκB release cytoplasmic NF-κB and permit its translocation to the nucleus, where it drives transcription of NF-κB-dependent genes. IKBKG mutations inhibit IκB phosphorylation and hence also impair nuclear translocation of NF-κB and expression of NF-κB-responsive genes, such as the ectodysplasin receptor responsible for the EDA phenotype, as well as UNG and AID, resulting in the immunologic abnormalities seen in these patients [43].
In addition, activation of CD40- and TLR-dependent pathways in dendritic cells and macrophages is impaired in patients with EDA-ID [41, 43]. Some of the patients present with hypogammaglobulinemia, but most fail to mount normal specific antibody production [43]. Defects in T and NK cells were also reported in HIGM6 [35, 41, 43]. This results in severe susceptibility to bacterial and mycobacterial infections in almost all patients with IKBKG mutations.
On the other hand, NF-κB is also involved in regulating the inflammatory process, as shown in patients with NOD2 and DLG5 mutations that are associated with Crohn’s disease [44] and by evidence that TLR signaling may be involved in the pathophysiology of autoimmunity [45]. Consequently, patients with IKBKG mutations also present with significant autoimmune manifestations, including inflammatory bowel disease, arthritis, and autoimmune hemolytic anemia [43].
Furthermore, NF-κB2-deficient mice present with multiorgan infiltration of activated T cells and high levels of autoantibodies to multiple organs. This was partially attributed to a marked reduction in the number and function of specific mature medullary thymic epithelial cells, which play a critical role in the negative selection of self-reactive T-cell clones as well as in the role of NF-κB in the regulation of thymic expression of Aire [46, 47].
Hence, IKBKG mutations leading to disturbances in the regulation of the NF-κB signaling pathway result in inflammatory and autoimmune abnormalities through both central and peripheral immune-related processes as well as by affecting nonimmune cells and processes.
X-Linked Agammaglobulinemia
X-linked agammaglobulinemia (XLA) is considered the prototype of PIDs. It is caused by impaired B-cell development and function due to mutations in the BTK gene that encodes for a cytoplasmic tyrosine kinase [48, 49]. Patients present with hypogammaglobulinemia and severely decreased numbers of peripheral B cells, leading to recurrent bacterial infections [50].
B cells are blocked at the pro-to pre-B-cell differentiation stage in the bone marrow resulting from impaired signal transduction through the pre-B-cell receptor due to the lack of BTK function.
It is surprising that up to 15% of XLA patients have autoimmune manifestations such as arthritis, diabetes, hemolytic anemia, and scleroderma despite the pronounced immunoglobulin deficiency [51, 52]. Although manifestations such as arthritis or the myositis often seen in XLA patients may in fact be of infectious origin, it has been postulated that chronic inflammation due to subclinical and chronic infections significantly contributes to immune dysregulation. It should be noted that the autoimmune and inflammatory manifestations of XLA do not always respond to immunoglobulin replacement therapy that otherwise significantly reduces the frequency of infections. This supports the notion that other BTK-dependent but antibody-independent mechanisms may be involved in the pathophysiology of autoimmunity in XLA [53]. These may include abnormal signaling by TLR9 through NF-κB, RelA activation [54, 55], as well as deficits in dendritic cell populations [56]. Thus, XLA serves as an important model for the understanding of autoimmunity in the near absence of immunoglobulins.
Other PIDs
The remaining three PIDs are X-linked severe combined immunodeficiency, properdin deficiency, and XL-dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome). To date, no significant autoimmune manifestations have been described in association with these diseases; nevertheless, this may be the result of masking of the phenotype by early treatment due to the severity of X-linked severe combined immunodeficiency, the relatively narrow phenotype of properdin deficiency that includes mainly susceptibility to fulminant meningococcal infections, and the extreme rareness of XL-dyskeratosis congenita [4••].
Conclusions
Our understanding of many aspects of the immune system has evolved through the ongoing research into PIDs. These unique “experiments of nature” enabled scientists to segregate the critical role of single gene products in the development, function, and homeostasis of the immune system. More recent observations of significant autoimmune phenomena associated with PIDs have led to further investigation of the role of PID-causing genes in the pathophysiology of more complex autoimmune disorders.
The group of X-linked PIDs is of particular interest due to the higher prevalence of autoimmunity in females and the growing body of evidence pointing to the distinctive role played by genes encoded on the X chromosome in autoimmunity and immune dysregulation. This is further reinforced by the high prevalence of significant autoimmune manifestations observed in most X-linked PIDs.
The study of female carriers of X-linked PID is also evolving and serves as a unique opportunity to study effects such as gene dosage and patterns of X chromosome inactivation that may play a role in autoimmunity. This has been demonstrated by various studies such as the studies of autoimmune phenomena in female carriers of X-CGD described previously [28, 30], as well as in recent studies of FOXP3 selective activation in CD4+CD25hi Tregs of healthy female carriers of different FOXP3 mutations [57]. Another approach has used the X chromosome monosomy seen in Turner’s syndrome to study the role of X-linked FOXP3 in the autoimmune susceptibility [58].
In combination, these approaches present an exceptional vantage point to the understanding of autoimmunity and its unique link to the X chromosome.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Carneiro-Sampaio M, Coutinho A: Tolerance and autoimmunity: lessons at the bedside of primary immunodeficiencies. Adv Immunol 2007, 95:51–82.
Notarangelo LD, Gambineri E, Badolato R: Immunodeficiencies with autoimmune consequences. Adv Immunol 2006, 89:321–370.
• Invernizzi P, Pasini S, Selmi C, et al.: Female predominance and X chromosome defects in autoimmune diseases. J Autoimmun 2009, 33:12–16. This review addresses the yet-to-be-answered enigma of the higher prevalence of autoimmune diseases in females by thoroughly analyzing relevant biological phenomena such as X chromosome inactivation, microchimerism, the role of sex hormones, and X chromosome monosomies.
•• International Union of Immunological Societies Expert Committee on Primary Immunodeficiences; Notarangelo LD, Fischer A, Geha RS, et al.: Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol 2009, 124:1161–1178. This report provides the updated classification of PIDs that was compiled by the International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies in June 2009. It includes novel forms of PID that have been recently discovered and additional pathophysiology mechanisms that account for PID in humans that have been unraveled.
Torgerson TR, Ochs HD: Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol 2007, 120:744–750.
Lyon MF, Peters J, Glenister PH, et al.: The scurfy mouse mutant has previously unrecognized hematological abnormalities and resembles Wiskott-Aldrich syndrome. Proc Natl Acad Sci U S A 1990, 87:2433–2437.
Chatila TA, Blaeser F, Ho N, et al.: JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic dysregulation syndrome. J Clin Invest 2000, 106:R75–R81.
Brunkow ME, Jeffery EW, Hjerrild KA, et al.: Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 2001, 27:68–73.
Fontenot JD, Gavin MA, Rudensky AY: Foxp3 programs the development and function of CD4 + CD25+ regulatory T cells. Nat Immunol 2003, 4:330–336.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M: Regulatory T cells and immune tolerance. Cell 2008, 133:775–787.
Rao A, Kamani N, Filipovich A, et al.: Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood 2007, 109:383–385.
Torgerson TR, Linane A, Moes N, et al.: Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology 2007, 132:1705–1717.
Caudy AA, Reddy ST, Chatila T, et al.: CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol 2007, 119:482–487.
Notarangelo LD, Miao CH, Ochs HD: Wiskott-Aldrich syndrome. Curr Opin Hematol 2008, 15:30–36.
Thrasher AJ, Burns SO: WASP: a key immunological multitasker. Nat Rev Immunol 2010, 10:182–192.
Albert MH, Bittner TC, Nonoyama S, et al.: X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood 2010, 115:3231–3238.
Devriendt K, Kim AS, Mathijs G, et al.: Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet 2001, 27:313–317.
Adriani M, Aoki J, Horai R, et al.: Impaired in vitro regulatory T cell function associated with Wiskott-Aldrich syndrome. Clin Immunol 2007, 124:41–48.
Marangoni F, Trifari S, Scaramuzza S, et al.: WASP regulates suppressor activity of human and murine CD4(+)CD25(+)FOXP3(+) natural regulatory T cells. J Exp Med 2007, 204:369–380.
Ma CS, Nichols KE, Tangye SG: Regulation of cellular and humoral immune responses by the SLAM and SAP families of molecules. Annu Rev Immunol 2007, 25:337–379.
Nichols KE, Ma CS, Cannons JL, et al.: Molecular and cellular pathogenesis of X-linked lymphoproliferative disease. Immunol Rev 2005, 203:180–199.
Rigaud S, Fondaneche MC, Lambert N, et al.: XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 2006, 444:110–114.
Crotty S, Kersh EN, Cannons J, et al.: SAP is required for generating long-term humoral immunity. Nature 2003, 421:282–287.
Holland SM: Chronic granulomatous disease. Clin Rev Allergy Immunol 2010, 38:3–10.
Rosenzweig SD, Holland SM: Phagocyte immunodeficiencies and their infections. J Allergy Clin Immunol 2004, 113:620–626.
Kang EM, Malech HL: Advances in treatment for chronic granulomatous disease. Immunol Res 2009, 43:77–84.
Kang EM, Choi U, Theobald N, et al.: Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood 2010, 115:783–791.
Cale CM, Morton L, Goldblatt D: Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol 2007, 148:79–84.
De Ravin SS, Naumann N, Cowen EW, et al.: Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol 2008, 122:1097–1103.
Schäppi M, Jaquet V, Belli D, Krause K-H: Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin Immunopathol 2008, 30:255–271.
Bleesing JJ, Souto-Carneiro MM, Savage WJ, et al.: Patients with chronic granulomatous disease have a reduced peripheral blood memory B cell compartment. J Immunol 2006, 176:7096–7103.
George-Chandy A, Nordstrom I, Nygren E, et al.: Th17 development and autoimmune arthritis in the absence of reactive oxygen species. Eur J Immunol 2008, 38:1118–1126.
Sanford AN, Suriano AR, Herche D, et al.: Abnormal apoptosis in chronic granulomatous disease and autoantibody production characteristic of lupus. Rheumatology (Oxford) 2006, 45:178–181.
Notarangelo LD, Lanzi G, Peron S, Durandy A: Defects of class-switch recombination. J Allergy Clin Immunol 2006, 117:855–864.
Jesus A, Duarte A, Oliveira J: Autoimmunity in hyper-IgM syndrome. J Clin Immunol 2008, 28:62–66.
Etzioni A, Ochs HD: The hyper IgM syndrome—an evolving story. Pediatr Res 2004, 56:519–525.
Jain A, Atkinson TP, Lipsky PE, et al.: Defects of T-cell effector function and post-thymic maturation in X-linked hyper-IgM syndrome. J Clin Invest 1999, 103:1151–1158.
White AJ, Withers DR, Parnell SM, et al.: Sequential phases in the development of Aire-expressing medullary thymic epithelial cells involve distinct cellular input. Eur J Immunol 2008, 38:942–947.
Kumanogoh A, Wang X, Lee I, et al.: Increased T cell autoreactivity in the absence of CD40-CD40 ligand interactions: a role of CD40 in regulatory T cell development. J Immunol 2001, 166:353–360.
Herve M, Isnardi I, Ng YS, et al.: CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J Exp Med 2007, 204:1583–1593.
Jain A, Ma CA, Liu S, et al.: Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol 2001, 2:223–228.
Zonana J, Elder ME, Schneider LC, et al.: A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet 2000, 67:1555–1562.
Orange JS, Levy O, Geha RS: Human disease resulting from gene mutations that interfere with appropriate nuclear factor-kappaB activation. Immunol Rev 2005, 203:21–37.
Friedrichs F, Henckaerts L, Vermeire S, et al.: The Crohn’s disease susceptibility gene DLG5 as a member of the CARD interaction network. J Mol Med 2008, 86:423–432.
Toubi E, Shoenfeld Y: Toll-like receptors and their role in the development of autoimmune diseases. Autoimmunity 2004, 37:183–188.
Zhang B, Wang Z, Ding J, et al.: NF-kappaB2 is required for the control of autoimmunity by regulating the development of medullary thymic epithelial cells. J Biol Chem 2006, 81:38617–38624.
Zhu M, Chin RK, Christiansen PA, et al.: NF-kappaB2 is required for the establishment of central tolerance through an Aire-dependent pathway. J Clin Invest 2006, 116:2964–2971.
Tsukada S, Saffran DC, Rawlings DJ, et al.: Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72:279–290.
Vetrie D, Vorechovsky I, Sideras P, et al.: The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361:226–233.
Winkelstein JA, Marino MC, Lederman HM, et al.: X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore) 2006, 85:193–202.
Howard V, Greene JM, Pahwa S, et al.: The health status and quality of life of adults with X-linked agammaglobulinemia. Clin Immunol 2006, 118:201–208.
Verbruggen G, De Backer S, Deforce D, et al.: X linked agammaglobulinaemia and rheumatoid arthritis. Ann Rheum Dis 2005, 64:1075–1078.
Knight AK, Cunningham-Rundles C: Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmun Rev 2006, 5:156–159.
Kubo T, Uchida Y, Watanabe Y, et al.: Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity. J Exp Med 2009, 206:1971–1982.
Lee KG, Xu S, Wong ET, et al.: Bruton’s tyrosine kinase separately regulates NFkappaB p65RelA activation and cytokine interleukin (IL)-10/IL-12 production in TLR9-stimulated B Cells. J Biol Chem 2008, 283:11189–11198.
Yong PF, Workman S, Wahid F, et al.: Selective deficits in blood dendritic cell subsets in common variable immunodeficiency and X-linked agammaglobulinaemia but not specific polysaccharide antibody deficiency. Clin Immunol 2008, 127:34–42.
Di Nunzio S, Cecconi M, Passerini L, et al.: Wild-type FOXP3 is selectively active in CD4 + CD25hi regulatory T cells of healthy female carriers of different FOXP3 mutations. Blood 2009, 114:4138–4141.
Su MA, Stenerson M, Liu W, et al.: The role of X-linked FOXP3 in the autoimmune susceptibility of Turner syndrome patients. Clin Immunol 2009, 131:139–144.
Disclosure
No potential conflict of interest relevant to this article was reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pessach, I.M. The Relationship of X-Linked Primary Immune Deficiencies and Autoimmunity. Curr Allergy Asthma Rep 10, 311–319 (2010). https://doi.org/10.1007/s11882-010-0127-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11882-010-0127-x