
Abstract
This study aims to investigate the concentrations and distributions of particulate matters and particle-bound polycyclic aromatic hydrocarbons (p-PAHs) in different inhalable fractions during haze periods in Phayao Province, northern Thailand and identify their possible emission sources through the characteristic ratios of selected PAHs, then determine an estimate of the potential lung cancer risk. The particulate samples were collected during haze periods between February 18 and April 8, 2014 and non-haze period from June 17 to 22, 2014 using an eight-stage cascade impactor. Each filter sample was weighed, ultrasonically extracted with acetonitrile, and then analyzed by SIM-GC-MS to identify 16 PAHs. The experimental results show that the concentrations of ultrafine (d ae < 0.4 μm), fine (0.4 μm < d ae < 2.1 μm), and coarse (d ae > 2.1 μm) particles during haze periods were in the range of 14.98–26.52 μg m−3, 62.57–101.52 μg m−3, and 55.27–89.68 μg m−3, respectively; whereas, their concentrations of 16 PAHs were in the range of 7.82–36.06 ng m−3, 26.66–61.89 ng m−3, and 9.35–30.93 ng m−3, respectively. The p-PAH distribution profiles during the haze periods were bimodal in coarse and accumulation modes, which are closely related to their particle size distributions. The characteristic ratios of BaP/BgP and INP/(BgP+InP) adsorbed on ultrafine and fine particles were 0.81–0.88 and 0.38–0.86 and 0.54–0.57 and 0.44–0.52, respectively. The values of B[a]Peq observed during haze periods varied from 9.57 to 29.05 ng m−3. Estimated lifetime cancer risks during haze periods ranged from 8.324 to 25.27 additional cases per 10,000 people exposed, which is about 10 times higher than that during non-haze period.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over the past decades, thick particulate smoke has regularly covered throughout the area of northern Thailand between late February and early May, which was known as a haze period. A large amount of air pollutants, particularly small particles, is emitted into the environment by illegal open burning of agricultural residues, household wastes, and forests (Sirithian et al. 2017; Phoothiwut and Junyapoon 2013; Wiriya et al. 2013). During haze periods, these air pollutants are trapped near the ground for a few weeks due to temperature inversion. Its weather and basin-shaped topography play an important role in the formation of temperature inversion. Under stagnant air conditions, air pollutants accumulate in the lower atmosphere leading to increased concentrations (Phoothiwut and Junyapoon 2013; Pengchai et al. 2009) as can be seen from the levels of PM10 often exceeding the daily concentration of 120 μg m−3 allowed by Thai ambient air quality standard (PCD 2015). This deteriorated air quality caused adverse health effects, environmental impacts, visibility decline, and also indirect impact on economic disruptions (Tan et al. 2011; Yang et al. 2006). An increase in the number of patients with respiratory and cardiovascular diseases has been reported during these periods (Pengchai et al. 2009).
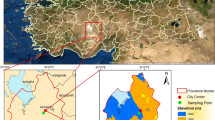
Phayao Province has a total area of about 6335.06 km2. Approximately 38% of the area is forest and around 24% of the area is devoted to agriculture. It is surrounded by high mountain ranges that have elevations from 300 to 1500 m above sea level. It is bordered by Chiang Rai to the north, Nan and Lao People’s Democratic Republic to the east, Phrae and Lampang to the south, and Lampang to the west as shown in Fig. 1 (PGO 2015). Like other provinces in the northern part of Thailand, open burning is a common way to clear agricultural residues after harvest. Furthermore, man-made forest fires have been regularly set to clear new land and harvest forest products. These activities introduce a large quantity of particulate matter, hydrocarbons, volatile organic compounds, and gases into the atmosphere which cause serious environmental problems.

Topographic map of Phayao Province and sampling location (Google Maps 2017)
Polycyclic aromatic hydrocarbons (PAHs) are typical compounds formed during incomplete combustion of organic substances. Some PAHs are potentially toxic or mutagenic to humans and other living organisms. Atmospheric PAHs are classified into two phases, vapor (g-PAHs) and particulate phases (p-PAHs), depending on their vapor pressures. Most of high molecular weight PAHs with low volatility tend to be adsorbed on particulate matter whereas low molecular weight PAHs with high volatility are mainly found in the vapor phase (Phoothiwut and Junyapoon 2013; Tan et al. 2011). It has been reported that the former PAHs have more carcinogenic potential than the latter ones (Keshtkar and Ashbaugh 2007). Previous studies also revealed that carcinogenic PAHs were likely correlated with small particulate matter (Lu et al. 2016; Samburova et al. 2016; Tan et al. 2011; Lee et al. 2008; Keshtkar and Ashbaugh 2007). Exposure to small particles during haze crisis over periods of time is associated with increased risk of developing lung cancer (IARC 2013). PAHs can be removed from the atmosphere by degradation, e.g., photolysis and chemical and biological degradations, as well as deposition. Particle-bound PAHs remain in the atmosphere for a long time because they cannot be readily degraded by photolysis and other pathways so they can be transported long distances from their original emission sources. Nowadays, Southeast Asian countries face the trans-boundary haze problem, thus reducing air pollutant emissions from open burning and forest fire which have become a regional concern (Shi et al. 2014; Huang et al. 2013).
Atmospheric particles consist of both primary particles, which are released directly into the atmosphere in solid or liquid form, and secondary particles, which are formed in the air by condensation processes of gases or chemical reactions. They contain a wide range of particle sizes and different compositions depending on the type of emission sources, weather conditions, chemical reactions, etc. The ratios and compositions of individual particles are continuously changed by either formation or destruction processes during their lifetimes. Hence, size distribution of particles and their compositions can reflect their emission sources as well as fate and transport in the environment (Lv et al. 2016; Curtius 2009; Lee et al. 2008). Several research have examined on the distribution of small particulates during haze periods because they can penetrate deeply into the lungs; however, there are few observations concerning potentially toxic components bound on these particles. Our previous study investigated on size distribution of particles and p-PAHs during haze periods in Lampang Province, Thailand. It was found that p-PAHs were predominantly detected on ultrafine particles; however, an estimate of the potential lung cancer risk was not observed. The objectives of this study were to examine the concentrations and distributions of particulate matter and p-PAHs in different inhalable fractions during haze periods (from February 18 to April 8, 2014) compared to those during non-burning period (between June 17 and 22, 2014) in Phayao Province, Northern Thailand. The effect of temporal variation on the distribution patterns of particulate matter and particle-bound PAHs during haze periods was observed. The characteristic ratios of selected PAHs were applied to determine source apportionment. An estimate of the potential lung cancer risk was also investigated.
Material and methods
Air sampling
The sampling site was located at the Observatory, Science and Astronomy Learning Center, Wieng Subdistrict, Muang District, Phayao Province, northern part of Thailand. It is centered at coordinates of latitude 19°10′6.7″ N and longitude 99°53′46″ E, with an elevation of 396 m above sea level (Fig. 1). Meteorological data including atmospheric temperature and pressure, relative humidity, wind speed, and wind direction were obtained from the monitoring station of the Meteorological Department of Thailand, located near the sampling site (TMD 2014). Numbers of hotspots in the region were also observed by Aqua satellite at 01.30 am, 10.30 am, 01.30 pm, and 10.30 pm, derived from the Department of National Parks, Wildlife and Plant Conservation, Thailand (DNP 2017).
To obtain enough sample due to the low concentrations of PAHs, air samples were continuously conducted for 120 h periods during haze episodes on February 18–23, 2014; February 24–March 1, 2014; March 11–16, 2014; March 17–22, 2014; and April 3–8, 2014. Before and after sample collections and the flow rates of sampling pump were measured 10 times repeatedly and then calculated for the average air volume. Paired t test was used to compare initial and final flow rates of the sampling pump that determined the blocking flow problem. During haze periods, the study area was covered with a blanket of smoke under stagnant atmospheric conditions. The air sample was also collected at the same site during the non-burning period on June 17–22, 2014. The particulate samples were collected on 81-mm-diameter quartz fiber filters (Toyo Roshi Kaisha, Japan) using a TISCH ambient eight-stage cascade impactor (Serial 436, Model 20-800, TISCH Environmental, USA) at a flow rate of 28.3l m−1 above the ground level of ~3 m. The particles were size-fractionated based on aerodynamic particle diameters in the following size intervals: 9.0–5.8, 5.8–4.7, 4.7–3.3, 3.3–2.1, 2.1–1.1, 1.1–0.7, 0.7–0.4, and <0.4 μm. Details of the air sampling method used in this study are described in Phoothiwut and Junyapoon (2013). The exposed filters were stored individually in sealed containers wrapped with aluminum foil and kept in plastic bags then immediately transported to the laboratory by keeping them in an ice box.
Analytical procedures
After sampling, the filter papers were equilibrated in a desiccator (Samplatec Corp, Germany) under controlled relative humidity (30–40%) and temperature (15–30 °C) for 24 h (US EPA 1999a) and then weighed on a five-digit electronic microbalance (Sartorius BA 210, France). After weighing, the sample filters were stored in the same containers and kept in a refrigerator at 4 °C for PAH analysis within 7 days. The particle concentrations were calculated by dividing the weight of the collected particulate matters by the volume of air sample adjusted to the standard ambient temperature and pressure (25 °C, 1 atm).
Sixteen PAHs consisting of naphthalene (Nap, 2-ring), acenaphthylene (Acy, 3-ring), acenaphthene (Ace, 3-ring), fluorene (Flu, 3-ring), phenanthrene (Phe, 3-ring), anthracene (Ant, 3-ring), fluoranthene (Fla, 4-ring), pyrene (Pyr, 4-ring), benzo[a]anthracene (BaA, 4-ring), chrysene (Chr, 4-ring), benzo[b]fluoranthene (BbF, 5-ring), benzo[k]fluoranthene (BkF, 5-ring), benzo[a]pyrene (BaP, 5-ring), dibenzo[a,h]anthracene (DbA,5-ring), indeno[1,2,3-cd]pyrene (InP, 6-ring), and benzo[ghi]perylene (BgP, 6-ring) were analyzed using the Compendium US EPA Method TO-13 (US EPA 1999b). Details of the analytical methods are explained in Phoothiwut and Junyapoon (2013). Briefly, each sample filter was cut into small pieces and placed in a 40 ml-glass amber vial. The PAH-containing filter was extracted with 15 ml of acetonitrile using ultrasonic agitation twice for 30-min periods (temperature <10 °C). Each sample extract was filtered by 0.45-μm pore size nylon filter and then concentrated by rotary evaporator (N-N series, Tokyo Rikakikai, Japan) under reduced pressure of 150 mbar at 30 °C. The cleanup procedure was applied by 3 ml (500 mg) Bond Elute C-18 cartridge (Varian, USA), which was conditioned with 20 ml of dichloromethane: n-hexane (1/1 v/v) before use. The concentrated sample was transferred onto the cartridge and then the wall of the vessel was rinsed twice with acetonitrile which was also added to the cartridge. Next, 5 ml of 20% dichloromethane in n-hexane was added to the cartridge with a flow rate of 3–5 ml min−1 to give fractions enriched in PAHs. The collected eluant from the cleanup procedure was reconcentrated to 1 ml by purging with ultrapure nitrogen (99.999% purity, Praxair, Thailand). All chemical reagents used were 99.5% HPLC grade. All samples were run in triplicate.
The 16 PAHs were determined by a HP-6890N gas chromatograph coupled to a HP-5973N mass spectrometer (Agilent Technologies, USA). One microliter of each sample was injected onto a HP-5MS GC fused-silica capillary column, 30 m × 0.25 mm i.d. × 0.25 μm film thickness, 5% diphenyl/95% dimethylsiloxane copolymer (J&W Scientific, USA). Helium (99.999% purity, Praxair, Thailand) was used as a carrier gas at a flow rate of 1.0 ml min−1. The inlet mode was splitless. The GC oven was programmed from 80 °C (initial temperature), holding at 80 °C for 2 mins, to 150 °C at 20 °C min−1, holding at 150 °C for 10 mins, then increased from 150 °C to 285 °C at 5 °C min−1, and then increased from 285 to 300 °C at 3 °C min−1, holding at 300 °C for 7 min. The transfer line between the GC and MS was operated at 300 °C. The MS was operated in electron impact (EI) mode at 70 eV with a mass range of 30–500 amu and a filament/multiplier delay of 6 min. The MS analysis was performed using the selected ion monitoring (SIM) mode. Chromatograms of samples were identified by comparison with retention time matching and fragmentation profiles against a mixture of 16 EPA PAH standards in CH2Cl2 (610 PAH Calibration Mix A, Restex, USA) and comparison of mass spectra obtained from the NIST mass spectral library database. Individual PAH was quantified relative to peak area of the 16 PAH standards. Recovery efficiencies were determined in triplicate using spike method. Detection limits of the analytical methods were based on three times the signal to noise ratio. Solvent and field blank samples were measured using the same method as the samples. All data were corrected with reference to the blanks, but were not corrected for recovery factors.
Calculation of lifetime lung cancer risk of PAHs
In this study, lifetime lung cancer risk of PAHs expressed as cancer risk per person exposed (ECR) from inhalation of PAHs bound on particles was estimated using the technique provided by the Office of Environmental Health Hazard Assessment (OEHHA) of the California Environmental Protection Agency (Cal EPA) (OEHHA 1993, 2005). The B[a]Peq was calculated by multiplying the concentrations of the ith PAH congener bound on each particle size range (C i) with the toxicity equivalency factor of the ith PAH congener (TEFi). Most of the TEF values used in this study were proposed by Larsen and Larsen (1998) except those of naphthalene (Nap), acenaphthylene (Acy), and acenaphthene (Ace) which were provided by Nisbet and LaGoy (1992). The total B[a]Peq was calculated by the sum of B[a]Peq for each PAH as listed in Eq. 1.
ECR is defined as the product of ∑B[a]Peq and URB[a]P as shown in Eq. 2, where URB[a]P (unit risk) represents the number of people at risk of contracting cancer from inhalation at a B[a]P equivalent concentration of 1 ng m−3 within a lifetime of 70 years. URB[a]P = 8.7 × 10−5 provided by the World Health Organization was used in this research (WHO 2000).
Results and discussion
Concentrations and size distributions of atmospheric particulate matters
Average 24-h concentrations of inhalable particle size fractions during haze and non-burning periods in Phayao Province are illustrated in Table 1. The average 24-h concentrations of particulate matter less than 10 μm (PM10) during haze periods varied from 132.82 to 215.50 μg m−3 with an average of 162.75 μg m−3, which exceeds the Thai ambient air quality standard of 120 μg m−3 (PCD 2017) and WHO Air Quality Guidelines of 50 μg m−3 (WHO 2000). They were about 30 times higher than that during non-haze period (5.34 μg m−3). These results are strongly associated with the concentrations of PM10 obtained from the Observatory, Science and Astronomy Learning Center monitoring station of the Pollution Control Department, Thailand at the same time (PCD 2015). The average 24-h concentrations of particulate matter less than 2.1 μm (PM2.1) during haze periods varied from 77.55 to 125.82 μg m−3 with an average of 93.36 μg m−3, which exceeds the Thai ambient air quality standard of 50 μg m−3 (PCD 2017) and WHO Air Quality Guidelines of 25 μg m−3 (WHO 2000). They were about 40 times higher than that during non-haze period (2.38 μg m−3). These results indicate that small particle concentrations during haze periods were significantly higher than those during non-burning periods, which are highly related to the numbers of hotspots detected throughout the region (Table 2). High concentrations of particulate matters were largely detected in March which was related to the burning activities indicated by the presence of hotspots. Weather conditions obtained from the Thai Meteorological Department are presented in Table 2. During haze periods, wind direction mainly blew from the north, therefore some small particulate matter may move from Chiang Rai Province to Phayao Province. However, wind speeds ranging from calm to light air could not effectively disperse the particles, resulting in increased concentrations. Furthermore, low relative humidity during haze periods led to decreased particle agglomeration which reduces deposition rate of the particles from the air which is consistent with the results of Lv et al. (2016). It was found that the atmospheric temperature and pressure during haze periods and non-burning period were not different.
In this study, eight inhalable fractions of these particles were classified into three size ranges according to their mechanism of formation: ultrafine particles (nuclei mode, d ae < 0.4 μm), fine particles (accumulation mode, 0.4 μm < d ae < 2.1 μm), and coarse particles (d ae > 2.1 μm). The concentrations of ultrafine, fine, and coarse particles in the atmosphere during haze periods were in the range of 14.98–26.52 μg m−3 with an average of 19.93 μg m−3, 62.57–101.52 μg m−3 with an average of 73.43 μg m−3, and 55.27–89.68 μg m−3 with an average of 69.39 μg m−3, respectively, which is about 44, 38, and 23 times of those during the non-haze periods (0.45, 1.93, and 2.96 μg m−3), respectively. These results show that fine (45.12% of the total mass) and coarse particles (42.64% of the total mass) were two major atmospheric particle components during haze periods, followed by ultrafine particles (12.24% of the total mass). Coarse particles, i.e., unburned hydrocarbons, likely resulted from incomplete combustion of organic matter while the fine particles may be transformed from ultrafine particles through the accumulation mechanism and condensation of vapor species (Lv et al. 2016). Moreover, these fine particles may be transported long distances from the adjacent provinces. These results do not correlate with those of our previous work (Phoothiwut and Junyapoon 2013), which found that the main fraction during haze periods was fine mode (42.9% of the total mass) followed by ultrafine mode (37.8% of the total mass) and coarse mode contributed only a minor fraction (19.3% of the total mass). This is probably because of the effects of meteorology and chemical reactions in the atmosphere on particle formation. In this study, coarse particles were the most abundant mode (55.43% of the total mass) during non-burning period followed by fine (36.14% of the total mass) and ultrafine (8.43% of the total mass) modes. These results reveal that the emission sources during haze periods and non-burning period were likely different.
The size distribution plots of atmospheric particles during haze and non-burning periods are shown in Fig. 2. The particle size distributions were bimodal during haze periods. The patterns of particle size distributions on February 18–23, February 24–March 1, March 11–16, and April 3–8, 2014 had a dominant coarse mode in the size range of 5.8–4.7 μm and a slightly less abundant mode in accumulation mode (1.1–0.7 μm range) except on March 17–22, 2014 which had a slight difference in accumulation mode at the size range of 0.7–0.4 μm. Unimodal distribution with a major peak in the coarse mode (5.8–4.7 μm range) was found during non-burning period. The patterns of particle size distributions during haze periods measured at different times were similar indicating that these particles tended to be derived from the same emission sources (Lv et al. 2016). A paired-sample t test was also conducted to compare flow rates before and after taking samples. There was no significant difference in the flow rates before (M = 28.346, SD = 0.037) and after (M = 28.123, SD = 0.378) taking samples. 95% CI for mean difference, −0.154 to 0.601; t(5) = 1.52, p = 0.189. These results indicate that pump flow was not blocked after taking samples for 120 h.

The size distributions of atmospheric particles during haze and non-haze periods in Phayao Province
Concentrations and size distributions of p-PAHs
Concentrations of total 16 p-PAHs during haze and non-burning periods are listed in Table 3. In this study, recovery efficiencies of 16 PAHs (n = 3) were shown in Table 4. The concentrations of total PAH adsorbed on ultrafine, fine, and coarse particles during haze periods were in the range of 7.82–36.06 ng m−3, 26.66–61.89 ng m−3, and 9.35–30.93 ng m−3, respectively. Their average concentrations were 23.01, 46.89, and 22.50 ng m−3, respectively, which are around 89, 35, and 10 times of those during non-burning period (0.26, 1.36, and 2.29 ng m−3), respectively. However, the daily concentrations of PAHs during haze periods did not exceed an average 8-h permissible exposure limit of the Occupational Safety and Health Administration (OSHA-PEL) of 200 ng m−3 (ATSDR 2017). These results indicate that total 16 PAHs were dominantly bound on fine particles (50.75% of the total PAHs) during haze periods followed by ultrafine particles (24.9% of the total PAHs) which is close to coarse particles (24.35% of the total PAHs) except those on April 3–8, 2014 which were significantly different in coarse, fine, and ultrafine particles, accounting for 45.76, 39.44, and 14.80%, respectively. This study is in a good agreement with other studies (Hazarika and Srivastava 2017; Lv et al. 2016; Phoothiwut and Junyapoon 2013; Lee et al. 2008; Keshtkar and Ashbaugh 2007). During non-burning period, total 16 PAHs were largely bound on coarse particles (approx. 58.57% of the total PAHs) followed by fine (about 34.78% of the total PAHs) and ultrafine particles (around 6.65% of the total PAHs), respectively.
Figure 3 shows that benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF), benzo[a]pyrene (BaP), dibenzo[a,h]anthracene (DbA), indeno[1,2,3-cd]pyrene (InP), and benzo[ghi]perylene (BgP), regarded as high molecular weight PAHs, were the main components of total PAHs found during haze periods. These results correlated with those of the work of Wiriya et al. (2016) indicating that high molecular weight PAHs were likely emitted from biomass burning. During non-burning period, the dominant PAHs found was napthalene (Nap) followed by benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF), and benzo[ghi]perylene (BgP), respectively. The concentrations of p-PAHs were much higher during haze periods than those during non-haze period. In this study, concentrations of Nap in particle phase during haze periods were found to be about twice higher than those during non-haze period. Although naphthalene is the most volatile PAH, several studies reported that it has been found in the particle phase (Hazarika and Srivastava 2017; Lv et al. 2016; Pavagadhi et al. 2013). This may be because it was adsorbed on the surface areas of small particles.

Concentrations of 16 PAHs bound on particles in inhalable fractions in Phayao Province. a During haze period. b During non-burning period
In this study, 16 PAHs bound on particles of different sizes were classified into four groups depending on the numbers of aromatic rings in their structures. During haze periods, most of the size distributions of 3- to 6-ring PAHs were bimodal, which had two major peaks in coarse mode (5.8–4.7 μm range) and accumulation mode (1.1–0.7 μm or 0.7–0.4 μm range). These results closely correlated with those of the previous studies (Lv et al. 2016; Lee et al. 2008) indicating that p-PAHs in the accumulation mode grew from primary components, which were directly emitted into the atmosphere, through condensation and/or coagulation mechanisms, and p-PAHs in the coarse mode caused from their adsorption affinity to particle’s surfaces. During haze periods, the size distributions of p-PAHs in each period of time were likely similar; thus, the distributions of 3- to 6- ring PAHs on March 17–22, 2014 (Figs. 4, 5, 6, and 7) were used as a representative. The p-PAH distribution profiles during the haze periods, except those of 6-ring PAHs, are closely related to their particulate size distribution results (Fig. 2); however, those during non-haze period had no uniform pattern and indeno[1,2,3-cd]pyrene (InP) was not detected. It was found that the concentrations of 5- to 6-ring PAHs were higher than those of 3- to 4-ring PAHs during haze periods, which is associated with their volatility property. The increased number of aromatic rings of PAHs resulted in increased intensities of peak in accumulation mode. At the same time, intensities of peak in coarse mode decreased and almost disappeared in 6-ring PAHs. This is because less volatile PAH compounds preferentially condensed on fine particles and more volatile PAH species are inhibited on smaller particles due to the Kelvin effect (Hien et al. 2007; Keshtkar and Ashbaugh 2007; Allen et al. 1996). The findings are closely related to the work of Lv et al. (2016). It reveals that during haze periods, fine particles were largely adsorbed with high molecular weight PAHs, which can deeply penetrate through the respiratory tract.

The size distributions of 3-ring PAHs in Phayao Province. a During haze period. b During non-burning period

The size distributions of 4-ring PAHs in Phayao Province. a During haze period. b During non-burning period

The size distributions of 5-ring PAHs in Phayao Province. a During haze period. b During non-burning period

The size distributions of 6-ring PAHs in Phayao Province. a During haze period. b During non-burning period
The characteristic ratios of selected PAHs bound on the particles were calculated and compared with references from previous studies as illustrated in Table 5. During thick haze periods between February 24 and March 22, 2014, the characteristic ratios of BaP/BgP bound on ultrafine and fine particles during haze periods were 0.81–0.88 and 0.38–0.86, respectively, which are likely close to the results of our previous study obtained during haze periods (Phoothiwut and Junyapoon 2013). The characteristic ratios of InP/(BgP+InP) adsorbed on ultrafine and fine particles were 0.54–0.57 and 0.44–0.52, respectively, which are corresponding to those of the previous studies observed during rice straw burning and haze periods (Phoothiwut and Junyapoon 2013; Yang et al. 2006). The characteristic ratios of BaP/BgP and InP/(BgP+InP) bound on ultrafine and fine particles during haze period on February 18–23, 2014 were 1.94, 1.82, and 0.60, 0.83, respectively, which are correlated with those of the work of Yang et al. (2006). In addition, the characteristic ratios of InP/(BgP+InP) adsorbed on fine particles are also associated with the results of Hazarika and Srivastava (2017) indicating that the emission sources were derived from wood combustion and diesel vehicle. These characteristic ratios alone could not conclude what the main source of PAHs is; however, it proposed that the emission sources from rice straw burning, wood combustion, and diesel vehicles play an important role in the formation of PAHs during haze periods in this study. The observations also reveal that the characteristic ratios of BaP/BgP and InP/(BgP+InP) bound on coarse particles during haze periods were greatly different in each sampling period. This possibly results from coarse particles containing various compositions influencing on the adsorption affinity of PAHs. The characteristic ratios of BaP/BgP and InP/(BgP+InP) during non-haze period could not be detected because atmospheric components consisted of low concentrations of PAHs. However, other PAH emission sources are required to be identified in future work.
Potential lung cancer risk
Figure 8a shows that the atmospheric particles were mainly composed of 5-ring PAHs (46.39–57.32% of total PAHs, averagely accounting for 53.66%) during haze periods followed by 6-ring PAHs (14.30–23.89% of total PAHs, averagely accounting for 20.1%), 4-ring PAHs (10.86–21.95% of total PAHs, averagely accounting for 16.03%), and 3-ring PAHs (7.23–14.10% of total PAHs, averagely accounting for 10.21%), respectively, whereas those during non-haze period predominantly contained 5-ring PAHs (57.07% of total PAHs) followed by 3-ring PAHs (20.74% of total PAHs), 4-ring PAHs (16.17% of total PAHs), and 6-ring PAHs (6.02% of total PAHs), respectively. The fractions of 6-ring PAHs bound on the particles during haze periods were about three times higher than those during non-burning periods while the fractions of 3-ring PAHs adsorbed on the particles during haze periods were twice lower than those during non-burning periods. These are consistent with other studies (Keshtkar and Ashbaugh 2007) indicating that high molecular weight PAHs with five or more rings were mainly associated with fine particulate matters.

The ratios of a 3–6-ring PAHs and b carcinogenic PAHs bound on particles during haze and non-burning periods in Phayao Province
In this work, 16 PAHs were classified into three groups according to IARC (2010): benzo[a]pyrene, benz[a]anthracene, and dibenz[a,h]anthracene as probable human carcinogens (Group 2A) and benzo[b]fluoranthene, benzo[k]fluoranthene, and indeno[1,2,3-cd]pyrene as possible human carcinogens (Group 2B) and the rest of the components as Group 3+not available (NA). The fractions of PAHs in Group 2A, Group 2B, and Group 3+NA during haze periods were in the range of 22.47–28.17% with an average of 24.49%, 32.81–48.34% with an average of 43.61%, and 29.19–39.02% with an average of 31.9%, respectively, while those during non-burning periods were 4.57, 32.60, and 62.83%, respectively (Fig. 8b). Carcinogenic PAHs (Group 2A) during haze periods had concentrations about five times higher than those during non-burning periods.
The concentrations of total p-PAHs and B[a]Peq during haze periods were 48.14–123.99 ng m−3 with an average of 92.41 ng m−3 and 9.57–29.05 ng m−3 with an average of 21.41 ng m−3, respectively, whereas those of non-burning period were 3.92 and 0.18 ng m−3, respectively (Table 6). The B[a]Peq values during haze periods extremely exceeded the European Union’s annual average B[a]Peq standard (1 ng m−3) (EC 2001). The lifetime cancer risks during haze periods varied from 8.324 E-04 (8.324 additional cases per 10,000 people exposed) to 2.527 E-03 (2.527 additional cases per 1000 people exposed), with an average of 1.834 E-04 (1.834 additional cases per 10,000 people exposed). They were around 10 times higher than those of the non-burning period (1.594E-05, 1.594 additional cases per 100,000 people exposed). These results obviously indicate that exposure to small particles bound on carcinogenic PAHs during haze periods potentially causes lung cancer.
Conclusions
This study indicates that the atmospheric particles during the haze periods in Phayao Province contained approximately ultrafine 12.24% of total mass, fine 45.12% of total mass, and coarse particles 42.64% of total mass, whereas those during non-haze periods were 8.43% of total mass, 36.14% of total mass, and 55.43% of total mass, respectively. The particle size distributions were bimodal with main peaks in the accumulation and coarse modes during haze periods while it was unimodal distribution with a major peak in the coarse mode during non-haze periods. The fractions of 16 PAHs bound on ultrafine, fine, and coarse particles during the haze period were 24.9% of the total PAHs, 50.57% of the total PAHs, and 24.35% of the total PAHs, respectively, while those during the non-haze period were 6.65% of the total PAHs, 34.78% of the total PAHs, and 58.57% of the total PAHs, respectively. The concentrations of particles and 16 PAHs adsorbed on ultrafine, fine, and coarse particles during haze periods were substantially higher than those during non-haze period. The atmospheric PAHs during the haze period were bimodal with major peaks in coarse and accumulation modes, whereas those during non-haze period had no uniform distributions. The characteristic ratios of BaP/BgP bound on ultrafine and fine particles during haze periods were 0.81–0.88 and 0.38–0.86, respectively. Their characteristic ratios of InP/(BgP+InP) were 0.54–0.57 and 0.44–0.52, respectively. Carcinogenic PAHs (5- to 6-ring PAHs) during haze periods had significantly higher concentrations than those during non-haze period. It implies that rice straw burning was possibly a significant source of fine particles containing high molecular mass PAHs. The lifetime cancer risks during haze periods were in the range of 8.324 to 25.27 additional cases per 10,000 people exposed, which is about 10 times higher than those during non-haze periods (1.594 additional cases per 100,000 people exposed). It indicates that exposure to small particles during haze periods is related to increased risk of developing lung cancer in people living in this area.
References
Allen JO, Dookeran NM, Smith KA, Sarofim AF, Taghizadeh K, Lafleur A (1996) Measurement of polycyclic aromatic hydrocarbons associated with size-segregated atmospheric aerosols in Massachusetts. Environ Sci Technol 30:1023–1031
ATSDR (Agency for Toxic Substances and Diseases Registry) (2017) Standards and regulations for polycyclic aromatic hydrocarbons (PAHs). Available from: https://www.atsdr.cdc.gov/csem/csem.asp?csem=13&po=8. Accessed in March 2017
Curtius J (2009) Nucleation of atmospheric particles. Eur Phys J Conferences 1:199–209
DNP (Department of National Parks, Wildlife and Plant Conservation, Thailand) (2017) Available from: www.dnp.go.th/forestfire/hotspot/hotspotmap_2557.htm. Accessed in June 2017
EC (European Commission) (2001) Air quality standards. Available from: http://ec.europa.eu/environment/air/quality/standards.htm. Accessed in Feb 2015
Google Maps (2017) Available from: https://www.google.co.th/maps/@19.254324,100.2325716,9z. Accessed in March 2017
Hazarika N, Srivastava A (2017) Estimation of risk factor of elements and PAHs in size-differentiated particles in the National Capital Region of India. Air Qual Atmos Health 10:469–482
Hien TT, Thanh LT, Kameda TK, Takenaka N, Bandow H (2007) Distribution characteristics of polycyclic aromatic hydrocarbons with particle size in urban aerosols at the roadside in Ho Chi Minh City, Vietnam. Atmos Environ 41(8):1575–1586
Huang K, Fu JS, Hsu NC, Gao Y, Dong X, Tsay SC, Lam YF (2013) Impact assessment of biomass burning on air quality in Southeast and East Asia during BASE-ASIA. Atmos Environ 78:291–302
IARC (International Agency for Research on Cancer) (2010) Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. Monogr Eval Carcinog Risks Hum 92:765–771
IARC (International Agency for Research on Cancer) (2013) Air pollution and cancer. In: Straif K, Cohen A, Samet J (Eds) IARC Scientific Publications No. 161
Keshtkar H, Ashbaugh LL (2007) Size distribution of polycyclic aromatic hydrocarbon particulate emission factors from agricultural burning. Atmos Environ 41:2729–2739
Larsen JC, Larsen PB (1998) Chemical carcinogens. In: Herster RE, Harrison RM (eds) Air pollution and health. Royal Society of Chemistry, Cambridge, pp 33–56
Lee JY, Shin HJ, Bae SY, Kim YP, Kang C-H (2008) Seasonal variation of particle size distributions of PAHs at Seoul, Korea. Air Qual Atmos Health 1:57–68
Lu W, Yang L, Chen J, Wang X, Li H, Zhu Y, Wen L, Xu C, Zhang J, Zhu T, Wang W (2016) Identification of concentrations and sources of PM2.5-bound PAHs in North China during haze episodes in 2013. Air Qual Atmos Health 9:823–833
Lv Y, Li X, Xu TT, Cheng TT, Yang X, Chen JM, Iinuma Y, Herrmann H (2016) Size distributions of polycyclic aromatic hydrocarbons in urban atmosphere: sorption mechanism and source contributions to respiratory deposition. Atmos Chem Phys 16:2971–2983
Nisbet ICT, LaGoy PK (1992) Toxic equivalent factors (TEFs) for polycyclic aromatic hydrocarbons (PAHs). Regul Toxicol Pharmacol 16:290–300
OEHHA (Office of Environmental Health Hazard Assessment) (1993) Benzo[a]pyrene as a toxic air contaminant, in: part B. Health effects of Benzo[a]pyrene. California Environmental Protection Agency, Office of Environmental Health Hazard Assessment, Air Toxicology and Epidemiology Section, Berkeley
OEHHA (Office of Environmental Health Hazard Assessment) (2005) Air toxics hot spots program risk assessment guidelines, in: part II. Technical support document for describing available cancer potency factors. California Environmental Protection Agency, Office of Environmental Health Hazard Assessment, Air Toxicology and Epidemiology Section, Oakland
Pavagadhi S, Betha R, Venkatesan S, Balasubramanian R, Hande MP (2013) Physicochemical and toxicological characteristics of urban aerosols during a recent Indonesian biomass burning episode. Environ Sci Pollut Res 20:2569–2578
PCD (Pollution Control Department) (2015) Haze crisis report in Northern Thailand. Available from: www.aqnis.pcd.go.th/northhaze. Accessed in Jan 2015
PCD (Pollution Control Department) (2017) Air quality and noise standard. Available from: www.pcd.go.th/info_serv/reg_std_airsnd01.html. Accessed in March 2017
Pengchai P, Chantara S, Sopajaree K, Wangkarn S, Tengcharoenkul U, Rayanakorn M (2009) Seasonal variation, risk assessment and source estimation of PM10 and PM10-bound PAHs in the ambient air of Chiang Mai and Lumphun, Thailand. Environ Monit Assess 154:197–218
PGO (Phayao’s Governor Office) (2015) Available from: www.phayao.go.th. Accessed in Febr 2015
Phoothiwut S, Junyapoon S (2013) Size distribution of atmospheric particulates and particulate-bound polycyclic aromatic hydrocarbons and characteristics of PAHs during haze period in Lampang Province, Northern Thailand. Air Qual Atmos Health 6:397–405
Samburova V, Connolly J, Gyawali M, Yatavelli RLN, Watts AC, Chakrabarty RK, Zielinska B, Moosmüller H, Khlystov A (2016) Polycyclic aromatic hydrocarbons in biomass-burning emissions and their contribution to light absorption and aerosol toxicity. Sci Total Environ 568:391–401
Shi Y, Sasai T, Yamaguchi Y (2014) Spatial-temporal evaluation of carbon emissions from biomass burning in Southeast Asia during the period 2001–2010. Ecol Model 272:98–115
Sirithian D, Thepanondh S, Laowagul W, Morknoy D (2017) Atmospheric dispersion of polycyclic aromatic hydrocarbons from open burning of agricultural residues in Chiang Rai, Thailand. Air Qual Atmos Health. doi:10.1007/s11869-017-0476-x
Tan J, Guo S, Ma Y, Duan J, Cheng Y, He K, Yang F (2011) Characteristics of particulate PAHs during a typical haze episode in Guangzhou, China. Atmos Res 102:91–98
TMD (Thai Meteorological Department) (2014) Meteorological information in 2014. Thai Meteorological Department, Ministry of Digital Economy and Society, Thailand
US EPA (Environmental Protection Agency) (1999a) Office of Research and Development, National Center for Environmental Assessment, Washington Office, Washington DC, EPA/600/P-99/002
US EPA (Environmental Protection Agency) (1999b) Compendium method TO-13A, determination of polycyclic aromatic hydrocarbons (PAHs) in ambient air using gas chromatography/mass spectrometry (GC/MS). Washington Office, Washington
WHO (World Health Organization) (2000) Air quality guidelines for Europe. 2nd edn. In: European series, No. 91 WHO regional publication, WHO Regional Office for Europe, Copenhagen, Denmark
Wiriya W, Chantara S, Sillapapiromsuk S, Lin NH (2016) Emission profiles of PM10-bound polycyclic aromatic hydrocarbons from biomass burning determined in chamber for assessment of air pollutants from open burning. Aerosol Air Qual Res 16:2716–2727
Wiriya W, Prapamontol T, Chantara S (2013) PM10-bound polycyclic aromatic hydrocarbons in Chiang Mai (Thailand): seasonal variations, source identification, health risk assessment and their relationship to air-mass movement. Atmos Res 124:109–122
Yang HH, Tsai CH, Chao MR, Su YL, Chien SM (2006) Source identification and size distribution of atmospheric polycyclic aromatic hydrocarbons during rice straw burning period. Atmos Environ 40(7):1266–1274
Acknowledgements
The authors are grateful to the Faculty of Science, King Mongkut’s Institute of Technology Ladkrabang for the financial support of this work. We also express our thanks to Observatory, Science and Astronomy Learning Center, Phayao Province for all the support provided during the sampling periods.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pooltawee, J., Pimpunchat, B. & Junyapoon, S. Size distribution, characterization and risk assessment of particle-bound polycyclic aromatic hydrocarbons during haze periods in Phayao Province, northern Thailand. Air Qual Atmos Health 10, 1097–1112 (2017). https://doi.org/10.1007/s11869-017-0497-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11869-017-0497-5