
Opinion Statement
Endocrine treatment resistance eventually develops during adjuvant and even more often during hormonal treatment for advanced breast cancer (ABC). An ESR1 gene mutation, which encodes for the estrogen receptor (ER) protein, is one of the potential mechanisms of therapy resistance. The ESR1 mutations result in conformational changes in the ER leading to subsequent estrogen-independent transcriptional activity. These mutations are found at a lower level in early stage when compared to metastatic BC, more often through selective pressure after aromatase inhibitor (AI) treatment. Recent studies have explored the role of ESR1 mutations as potential prognostic and predictive biomarkers and showed that ESR1 mutations are likely associated with a more aggressive disease. However, definitive associations with outcome in order to make a specific treatment recommendation are yet to be found. The development of targeted therapy directed to ESR1-mutated clones is an appealing concept, and preclinical and clinical works are in progress. ESR1 mutations represent an exciting field with a rapidly increasing number of recent publications that will likely advance the knowledge of treatment resistance mechanisms and pave the way into more individualized patient endocrine treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is a heterogeneous disease comprising distinct clinical, histopathological and molecular subtypes. Hormone-receptor-positive (HR+) tumors represent the most common form of breast cancer and account for most of the deaths from the disease. Endocrine therapy (ET) is recommended virtually to all patients with HR+ early-stage tumors and is the mainstay of treatment of HR+ advanced breast cancer (ABC). However, due to a diversity of mechanisms of resistance, approximately 20–30% of patients with early stage breast cancer will relapse during or after completion of adjuvant ET [1] and recurrences can sometimes occur years or decades after diagnosis despite curative-intent loco-regional and systemic therapies [2••]. In the metastatic setting, even though most patients derive benefit from initial ET, with disease stabilization or tumor shrinkage, development of resistance and disease progression invariably occurs [2••]. Breast tumors undergo molecular evolution over the course of the disease and especially during selective pressure by therapeutic strategies such as estrogen deprivation, with the acquisition of new alterations that confer resistance to therapy such as mutations of the ESR1 gene, which encodes the estrogen receptor (ER).
This article reviews published research and future perspectives regarding ESR1 mutations in breast cancer, addressing epidemiological and pathophysiological issues with conceivable clinical implications. The potential use of ESR1 mutational status as a prognostic and predictive biomarker, along with the development of therapeutic strategies targeting ESR1-mutated tumor will also be explored.
The ER Pathway
Estrogens (estradiol and estrone) play a pivotal role in the initiation and progression of HR+ breast cancer. Effects of estrogen are mediated through ER, a protein encoded by the ESR1 gene of the steroid hormone receptor superfamily that is expressed in approximately 70% of breast cancers. ER expression is one of the determinant characteristics in classifying breast cancer subtype and assigning therapeutic strategies. Experimental and clinical researches have established the central role of ER and its ligands in normal mammary gland development, and in the initiation and progression of breast tumors [3].
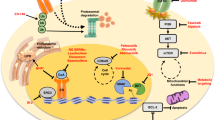
ER is predominantly a nuclear protein that functions as a ligand-dependent transcription factor. Functionally, the ER consists of two transcriptional activation domains: the ligand-dependent domain (AF-2) and the ligand-independent activation function domain (AF-1) [4]. The ligand-binding domain (LBD) is located in the C-terminal region, while the DNA-binding and hinge domains are found in the central core of the protein [2••]. Estrogen binding initiates a variety of events that activate the ER pathway and induces conformational changes in the LBD, allowing the estrogen-ER complex to interact with specific DNA sequences (estrogen response elements–EREs) while networking with co-activating and co-repressor proteins to regulate the transcription of estrogen-responsive genes that are influential in a variety of physiological and pathological processes [5••, 6,7,8].
Importantly, estrogens interact with high affinity with other cellular components, including membrane receptors. Some effects of estradiol, like activation of mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) pathways, appear to involve predominantly a direct action of constituents of the ER pathway at the plasma membrane rather than genomic modulation. The aberrant activation of growth factor receptors (GFRs), including human epidermal growth factor receptor (HER) family receptors [9], fibroblast growth factor receptor 1 (FGFR1) [10], and their downstream signaling components, including phosphoinositide 3 kinase–serine/threonine protein kinase (PI3K–AKT pathway) [11], has been associated with acquired ET resistance. These forms of “non-genomic” effect of the ER pathway are summarized in Fig. 1 and have been comprehensively reviewed elsewhere [2••, 6].

ER pathway and mechanisms of resistance. Estrogen-bound estrogen receptor (ER), in association with a variety of co-activators and co-repressors, exerts its classical genomic action as a transcription factor through the estrogen response element (ERE) of target genes. ER can also mediate an ERE-independent genomic effect via interaction with other transcription factors. In addition, ER can be activated via plasma membrane crosstalk with other growth factor receptor pathways that phosphorylate ER or its co-regulators. Reprinted from Ma C, Reinert T, Chmielewska I, and Ellis M., Mechanisms of aromatase inhibitors resistance. Nat Rev. Cancer 2015;15:261–75, with permission from Springer Nature.
Contemporary Endocrine Therapy Strategies
Beatson’s historical observations on the regression of breast cancer following oophorectomy more than a century ago provided the initial insight into the hormone-related nature of breast cancer. Targeting the ER pathway with endocrine agents could be considered the first molecularly targeted cancer therapy and remains the mainstay of treatment for all stages of HR-positive disease [12•]. The main strategies of ET include treatments that result in estrogen deprivation (e.g., ovarian ablation, aromatase inhibitors [AIs]), drugs that antagonize the ER (e.g., tamoxifen), and ER degraders (e.g., fulvestrant) [13].
Even though ET is associated with significant clinical benefits throughout all stages of the breast cancer clinical continuum, breast tumors are known to undergo genomic evolution, with the acquisition of new alterations that confer resistance to therapy. In the metastatic setting, most patients achieve benefit from initial ET, with symptoms palliation, disease stabilization, or tumor shrinkage. Nonetheless, subsequent lines of treatment result in shorter periods of response, denoting the development of resistance and disease progression that invariably occurs [2••].
Advances in the understanding of molecular interactions between the ER pathway and growth factor, and metabolic and cell-division pathways have been translated into clinical trials that showed improved outcomes using targeted therapies that modulate hormone signaling and interfere with resistance mechanisms [12•]. Therefore, the treatment algorithm for HR+ advanced breast cancer is evolving, and combinations of endocrine agents with targeted therapies that modulate endocrine resistance, such as mammalian target of rapamycin (mTOR) and cyclin-dependent kinase (CDK) 4/6 inhibitors, have been incorporated into clinical practice [12•, 13, 14]. Major paradigms that have been guiding clinical practice include the sequential use of endocrine agents and the indication of ET in all cases, except those with an impending visceral crisis or proven endocrine resistance. However, the lack of predictive biomarkers, tumor heterogeneity and limitations in the design of clinical studies make the development of a rational approach to the management of HR+ ABC challenging, especially in regard to the optimal sequencing strategy of therapeutic agents [12•, 15].
ESR1 Mutation: Physiopathology and Potential Role as a Prognostic and Predictive Biomarker
The initial description of ESR1 mutations in breast cancer patients dates back to 1997 by Zhang et al., which identified three missense mutations in a cohort of 30 tumors from metastatic patients [16]. Two of the three mutations were similar to wild-type ER; however, Y537N showed a potent estradiol-independent transcriptional activity, due to the induction of conformational changes in the ER. A previous preclinical report from Weis et al. showed similar results of the ligand-independent transcriptional activity of a different mutation, Y537S ER, in breast cancer cell lines, showing that the amino acid substitutions at the 537 position facilitate an active conformation of the ER in its active form [17].
Mutant ER recruits coactivators in the absence of hormone and confers antiestrogen resistance by altering the conformational dynamics of the loop connecting Helix 11 and Helix 12 in the LBD of ER that leads to a stabilized agonist state and an altered antagonist state that resists inhibition [18]. Functional studies showed that these mutated receptors have a decreased affinity for tamoxifen and estradiol [19].
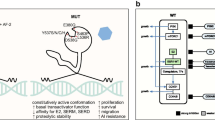
In the past few years, several other ESR1 mutations and genomic alterations leading to endocrine treatment resistance have been described. The majority of the ESR1 mutations are located in a hotspot region of the ligand-binding domain (LBD) comprising the codons 534 through 538, located mainly at two amino acids in the Y537N/C/S and D538G [20,21,22,23,24]. Another recurrent ESR1 LBD mutation, E380Q, has been described (Fig. 2) [20, 21, 24].

The ESR1 gene and its most common mutations. A schematic diagram of estrogen receptor-α (ERα) mutations and their frequencies in ER+ metastatic breast cancer after therapy with aromatase inhibitors (AIs) and other endocrine agents. The structural domains of ERα are shown, including the transcription activation function 1 (AF1) domain, the DNA-binding domain (DBD), the receptor dimerization and nuclear localization (hinge) domain, and the ligand-binding domain (LBD) and AF2 domain. Reprinted from Ma C, Reinert T, Chmielewska I, and Ellis M., Mechanisms of aromatase inhibitors resistance. Nat Rev. Cancer 2015;15:261–75, with permission from Springer Nature.
Recently, Martin et al. described for the first time the identification of naturally occurring ESR1 mutations in ESR1-positive BC cell lines. This study also supported the notion that activating mutations in the ESR1 are sufficient for driving acquired resistance that does not require changes in other signaling pathways. Of note, while Y537S was not inhibited by tamoxifen, it was inhibited by fulvestrant at current usual doses [25•].
Veeraraghavan et al. identified a recurrent rearrangement event between ESR1 and CCDC170 leading to a gain-of-function alteration. This abnormal form is associated with increased motility, tumorigenicity, and endocrine resistance [26]. Li et al. described another interesting rearrangement in which the C-terminal end of ESR1 is fused to the C-terminus of YAP1. The ESR1/YAP1 mutants showed hormone-independent growth and complete resistance to fulvestrant since the LBD is not present in this chimeric protein [20].
In primary breast cancer samples, these mutations are found in a very low frequency, being identified in only 0.5% of the 962 samples in The Cancer Genome Atlas set, using next-generation sequencing (NGS) [27]. However, the development of newer ultrasensitive technologies such as digital droplet PCR (ddPCR) challenged this finding. Using ddPCR in a cohort of 270 primary tumors, Takeshita et al. reported a 2.5% (7/270) prevalence of ESR1 mutations [28] and Wang et al. found an even higher prevalence of 7% (3/43) [29]. It is reasonable to hypothesize, based on these studies, that the prevalence of ESR1 mutations in primary tumors is higher than previously reported in NGS studies.
In contrast with primary ER-positive BC sequencing studies, the NGS of metastatic ER-positive BC samples showed a much higher prevalence of ESR1 mutations between 11 and 55%, especially in patients previously treated with an AI [20,21,22,23,24]. When more advanced technologies, such as sequencing of plasma ctDNA or ddPCR, were used to analyze ER-positive BC patients that progressed after an AI, the prevalence of ESR1 mutations ranged from 11 to 39% [30••, 31•, 32, 33••, 34, 35].
Although these mutations were first described in NGS studies analyzing stored metastatic tissue specimens, this method is not the most convenient for prospective studies, as it requires sequential biopsies that are invasive and, often, risky procedures [4]. In the search for a non-invasive method of detection of DNA mutations, Dawson et al. demonstrated in their seminal study the utility of serial evaluations of ctDNA in plasma samples of breast cancer patients [36]. In that study, ctDNA was detected in 97% of the metastatic patients who had a PIK3CA or TP53 mutation in their primary tumor specimens, ctDNA levels were correlated with changes in tumor burden and increasing ctDNA levels were correlated with worse prognosis. To identify and validate the occurrence of ESR1 mutations in prospective trials, several groups are studying non-invasive techniques to detect them in ctDNA isolated from plasma samples [28, 30••, 32, 33••, 37, 38]. In this setting, ddPCR seems to be a simpler and more sensitive approach than NGS [37,38,39]. In a series of studies, Takeshita et al. demonstrated the utility and feasibility of ddPCR monitoring of ESR1 mutations in ctDNA in metastatic BC patients [40], the clinical significance of on-treatment ESR1 mutations monitoring [41] and finally evaluated the independent distribution of ESR1 mutations between plasma and tumor tissue in 35 metastatic BC patients [42].
With the emerging knowledge of the presence of ESR1 mutations mainly in advanced and metastatic BC, researchers have been exploring its potential as a prognostic and predictive biomarker. Wang et al. found ESR1 mutations in 7 of 29 ctDNA samples of advanced ER-positive BC patients and showed that changes in ESR1 mutations detection and allele frequencies were associated with treatment [29]. In a cohort of 171 advanced BC patients, Schiavon et al. identified ESR1 mutations in 18 ER+ patients, all of them with prior exposure to treatment with an aromatase inhibitor. These patients had significantly lower PFS on subsequent AI-based therapy (HR = 3.7, 95%CI 1.9–76.9, p = 0.008). Moreover, the authors found a statistically significant difference in the detection of ESR1 mutations between patients who received an AI in the adjuvant setting and patients who received an AI in the metastatic setting (5.8 versus 36.4%, p = 0.0002) [33••]. The secondary analysis of the BOLERO-2 clinical trial that randomized 724 MBC patients to exemestane plus placebo versus exemestane plus everolimus had the objective to determine the prevalence of ESR1 mutations and whether they were associated with poor PFS [30••]. The authors identified mutations in 156 of 541 (28.8%) evaluable patients and showed a shorter OS in those patients when compared to WT ESR (WT 32.1 months, D538G 25.99 months, and Y537S 19.98 months). To further investigate the relationship between ESR1 mutations and survival, and given the fact that those mutations are rare in the primary setting, Fribbens et al. enrolled 83 patients on the first-line AI for MBC in a prospective study to collect plasma samples every 3 months to disease progression [43]. Twenty-two of 39 (56.4%) patients presented ESR1 mutations at progression and those mutations were detectable 6.7 months prior to clinical progression (95% CI 3.7-NA). Of those 22 patients, 9 presented polyclonal mutations (40.9%). In a retrospective study, Clatot et al. demonstrated similar results, where ER-positive MBC patients with ESR1 mutations had a decreased PFS (5.9 versus 7 months, p = 0.002) and OS (15.5 versus 23.8 months, p = 0.0006) than patients without detectable mutations [31•]. In this study, 75% of the patients had detectable mutations 3 months prior to clinical progression.
In the FERGI trial, a randomized phase 2 study that compared fulvestrant plus placebo versus fulvestrant plus pictilisib, ESR1 mutations were detected in 78 of 207 (37%) patients with MBC but they were not associated with differences in PFS. Decreases in allele frequencies of ESR1 mutations, however, were associated with clinical response in this trial [34]. In the retrospective analysis of the SOFEA and PALOMA-3 trials, seven ESR1 mutations were evaluated at baseline ctDNA samples of metastatic ER-positive BC patients that progressed on endocrine treatment [32]. In the SOFEA trial, ESR1 mutations were identified in 63 of 161 (39.1%) patients and in the PALOMA3 trial, such mutations were present in 91 of 360 (25.3%) patients. In the first, patients with ESR1 mutations had an improved PFS with fulvestrant when compared to exemestane (HR = 0.52, 95% CI 0.30–0.92, p = 0.02); in the latter, patients receiving a combination treatment with fulvestrant plus palbociclib showed improved PFS independent of ESR1 mutation status. As discussed above, ESR1 mutations are likely associated with more aggressive biology and poor PFS but definitive association with response to different treatment regimens is yet to be found. However, results from the clinical trials discussed above support that the dose of fulvestrant used might be sufficient to overcome treatment resistance induced by ESR1 mutations.
A recent publication by Toy et al. [44] analyzed the spectrum of ESR1m from over 900 patients. Seventy-eight ESR1m were detected in 10%, with D538G being the most frequent (36%), followed by Y537S (14%). A variety of novel-activating mutations were also identified (e.g., Y537D, V422del, L469V). While many mutations lead to constitutive ER activation and resistance to SERMs, only selected mutants such as Y537S caused significant changes associated with fulvestrant resistance in vitro. Accordingly, more potent and bioavailable SERDs inhibited tumors driven by Y537S more effectively than fulvestrant, whereas the inhibition was equivalent in tumors driven by D538G, E380Q, or S463P.
Patient and disease characteristics could be associated with an increased risk of development of ESR1m. Firstly, most patients with tumor-harboring ESR1m have experienced a protracted clinical course before the detection of the mutant clones. Some studies have reported an association between the frequency of hotspot LBD mutations and previous use of multiple lines of ET [5••], thereby supporting the concept that ESR1m is predominantly a mechanism of acquired (secondary) resistance that emerges after long-term ET [45]. Interestingly, kinetic studies reported that circulating ESR1m can be detected before clinical progression in 75% of cases [46]. If further confirmed and validated, these results may eventually lead to evaluating the strategy of earlier changes in therapy based on liquid biopsies, before clinical and radiological demonstrations of disease progression.
Secondly, ESR1m is mostly a mechanism of AI resistance rather than a non-specific mechanism of resistance to endocrine agents in general. ESR1m mutations have rarely been identified in patients only exposed to tamoxifen [47,48,49]. Even though analysis of tamoxifen resistance has contributed to the understanding of ligand-independent ER activity, it must be emphasized that while SERMs and AI resistance share many similar features, their effects on ER signaling are distinct [50].
Early reports of ESR1m in tumor samples have been obtained from different organ sites, including the skin, bone, lymph nodes, liver, and lungs, indicating that these mutations do not display specific organotropism [5••, 20, 21, 48, 51]. Contrastingly, multivariable analyses based on liquid biopsies of patients included in the PALOMA-3 and SOFEA studies showed that the detection of ESR1m is associated with bone and visceral disease and may suggest that ESR1m are rarely present in patients with locoregional recurrence only. [49]. These association needs to be evaluated in further studies.
Moreover, it is not clear if the frequency of ESR1m differs markedly between tumors exposed to AI during (neo)adjuvant therapy and those exposed in the metastatic setting. Studies demonstrating a higher frequency of mutant clones in advanced disease (compared to early stage disease) have hypothesized that preexisting ESR1m subclones are selected following estrogen deprivation, but the tumor burden in the micrometastatic setting might be too low for such clones to be detected in comparison with the higher tumor burden seen in overt macrometastatic disease [47, 52]. This hypothesis-generating association needs to be further evaluated in prospective studies, especially so with the increasing use of extended adjuvant ET [53].
Taken together, these evidences support that ESR1m is a mechanism of acquired ET resistance occurring in a significant proportion of AI refractory patients (see Table 1). ESR1m is clearly a prognostic biomarker consistently associated with statistically and clinically inferior survival outcomes (both PFS and OS) in comparison with patients with wild-type ESR1m. Additionally, ESR1m can be considered a predictive biomarker associated with inferior outcomes with further AI monotherapy. The impact of subsequent chemotherapy treatment has not been adequately studied. As previously discussed, the presence of ESR1m does not seem to have any impact on the benefit associated with mTOR or CDK4/6 combinations. It is not known how the increasing use of CDK4/6 inhibitors both in the first- and second-line treatment will impact the subsequent emergence of ESR1m.
ESR1 Mutation as a Therapeutic Target
The development of targeted therapy directed to ESR1-mutated clones is an appealing concept, and preclinical and clinical development of rationale-based novel therapeutic strategies that inhibit these ER mutants has the potential to substantially improve treatment outcomes. Efforts to develop new agents with superior bioavailability, pharmacokinetics, and potent antiestrogenic activity in the breast have led to the discovery and characterization of the second and third generations of SERDs, SERMs, and SERDs-SERMs hybrids that are being evaluated in phase I/II clinical trials following preclinical studies that demonstrated evidence of efficacious inhibition of ER-LBD mutants [5••, 54].
Among the most promising agents are the oral SERDs AZD9496 [55], ARN810 [56], GDC0810 [57], and RAD1901 [58]. Two antiestrogens with mixed SERM/SERD activity, bazedoxifene and pidendoxifene, have shown efficacy in preclinical models of ET resistance, with bazedoxifene demonstrating activity against a patient-derived xenograft (PDX) model expressing the ESR1 Y537S mutation [59].
The activity of mutated ER remains highly dependent on the interaction with ERE, especially recruitment of co-activators. New strategies with therapies targeting ER co-activators, such as small-molecule SRC-3 inhibitors, may offer another approach to target ESR1m and are being tested alone and in combination with ER antagonists [60].
The presence of ESR1m is associated with several different genetic and epigenetic alterations; therefore, testing of novel therapeutic approaches in preclinical models that include distinct molecular backgrounds is essential and highlights the potential of techniques like patient-derived xenografts (PDXs) and ex vivo cultures of circulating tumor cells (CTCs) from patients with metastatic ER+ breast cancer. Indeed, Yu et al. [61] tested inhibitors of multiple pathways, alone or in combination with endocrine agents and elicited the efficacy of shock protein 90 (HSP90), PI3K, and mTOR inhibitors in this context, especially in combination with fulvestrant. Targeting HSP 90, which is the chaperone protein of ER, might be useful to treat Y537S ESR1-mutated tumors. The authors showed that mutant ESR1 tumors are highly dependent on HSP90, and preclinical studies with the HSP90 inhibitor STA9090 demonstrated cytotoxicity alone and in combination with SERMs and SERDs to ex vivo-cultured circulating breast tumor cells [61]. Interestingly, the allele frequency of ESR1m was associated with the sensitivity to HSP90 inhibition.
Conclusion
The traditional clinicopathological features including grade, histological subtype, ER, PR, and HER2 status do not reflect the significant heterogeneity of ER-positive breast cancer. As previously discussed, estrogen-independent growth often exists de novo at diagnosis or develops during ET. The generation of clinically relevant predictive biomarkers to allow optimal sequencing of therapeutic strategies and to guide more individualized patient care is a research priority.
The discovery of recurrent ESR1 mutations within ER+ advanced breast cancer introduced new concepts for the understanding and modulation endocrine resistance [4]. Recent data analyzing ctDNA, which better captures the intra-tumoral heterogeneity, reported that the ESR1 mutations are found in approximately 40% of patients with AI-refractory disease [32, 62]. The use of liquid biopsies for assessing ESR1m is a promising tool for the continued investigation of the clinical role of these mutations as a predictive biomarker. However, this test will need to be standardized before being incorporated into clinical practice. Particularly, a standardized cutoff has not been established, and it is not known if the allele frequency is more important than a dichotomized result and if there are potential clinical implication related to the kinetics of the allele frequency based on sequential testing [63].
Integrative approaches using a variety of technologies and data, leading to comprehensive evaluations of gene expression, proteomics, and epigenetic regulators of the genome may be translated into biomarkers and therapies associated with clinical benefit for cancer patients [64]. Future research efforts may allow ESR1 mutational status to be prospectively tested as predictive biomarker, stratification factor, and as a therapeutic target.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Josefsson M, Leinster S. Aromatase inhibitors versus tamoxifen as adjuvant hormonal therapy for estrogen sensitive early breast cancer in post-menopausal women: meta-analyses of monotherapy, sequenced therapy and extended therapy. Breast. 2010;19:76–83.
•• Ma C, Reinert T, Chmielewska I, Ellis M. Mechanisms of aromatase inhibitors resistance. Nat Rev Cancer. 2015;15:261–75. Comprehensive review of mechanisms of resistance to AIs cancer considering both genomic and cell biological explanatations as to why ER+ breast cancer cells progress and cause an incurable systemic disease.
Huang B, Warner M, Gustafsson J. Estrogen receptors in breast carcinogenesis and endocrine therapy. Mol Cell Endocrin. 2014.
Reinert T, Saad ED, Barrios CH, Bines J. Clinical implications of ESR1 mutations in hormone receptor-positive advanced breast Cancer. Front Oncol. 2017;7:26.
•• Jeselsohn R, Buchwalter G, De Angelis C, et al. ESR1 mutations: a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. 2015;12:573–83. Very important publication that established the role of ESR1 as a mechanism of resistance present in AI-refractory patients but not as a mechanism of primary endocrine resistance.
Osborne CKSR. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–47.
Schiff RMS, Shou J, et al. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. 2004;10:331S–6S.
Rea S. Advanced concepts in oestrogen receptor biology and breast cancer endocrine resistance: implicated role of growth factor signalling and oestrogen receptor coregulators. Cancer Chemother Pharmacol. 2005;56:10–20.
Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–35.
Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–94.
Cavazzoni ABM, Fumarola C, et al. Overcoming acquired resistance to letrozole by targeting the PI3K/Akt/mTOR pathway in breast cancer cell clones. Cancer Lett. 2012;323:77–87.
• Reinert T, Barrios C. Optimal management of hormone receptor positive metastatic breast cancer in 2016. Ther Adv Med Oncol. 2015;7:304–20. Review of the literature that discusses advances and challenges in current treatment of ER+ advanced breast cancer. Issues about optimal sequencing of agents, definition of patterns of endocrine resistance, and factors that should be taken into account when choosing the ideal ET for the individual patient are discussed.
Reinert T, Barrios C. Overall survival and progression-free survival with endocrine therapy for hormone receptor-positive, HER2-negative advanced breast cancer: review. Ther Adv Med Oncol. 2017;9:693–709.
Cruz M, Reinert T, Cristofanilli M. Emerging innovative therapeutic approaches leveraging cyclin-dependent kinase inhibitors to treat advanced breast cancer. Clin Pharmacol Ther. 2017; https://doi.org/10.1002/cpt.965.
Rugo HSRB, Macrae E, Barton DL, Connoly HK, Dickler MN, et al. Endocrine therapy for hormone receptor–positive metastatic breast cancer: American Society of Clinical Oncology guideline. J Clin Oncol. 2016;34:3069–103.
Zhang QX, Borg A, Wolf DM, Oesterreich S, Fuqua SA. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997;57:1244–9.
Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol. 1996;10:1388–98.
Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. elife. 2016;5
Fanning S, Mayne C, Dharmajaran V, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. elife. 2016;5.
Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4:1116–30.
Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–45.
Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51.
Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-alpha: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013;73:6856–64.
Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–67.
• Martin L, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T, et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun. 2017;8:1–15. Recent data reporting naturally occuring ESR1m in cell lines implicating that a minor fraction of mutant clones may be present in the primary tumor and could be responsible for primary endocrine resistance.
Veeraraghavan J, Tan Y, Cao XX, et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun. 2014;5:4577.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, et al. Droplet digital polymerase chain reaction assay for screening of ESR1 mutations in 325 breast cancer specimens. Transl Res. 2015;166:540–53 e2.
Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, Watters RJ, et al. Sensitive detection of mono- and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast Cancer patients. Clin Cancer Res. 2016;22:1130–7.
•• Chandarlapaty S, Chen D, He W, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016;2:1310–5. Evaluation of ESR1m pattenrs in AI-refractory patients enrolled in the BOLERO2 trial. ESR1m are associated with both prognostic and predictive implications. Additionally, heterogeneity among mutant clones affecting different codons is described.
• Clatot F, Perdrix A, Augusto L, et al. Kinetics, prognostic and predictive values of ESR1 circulating mutations in metastatic breast cancer patients progressing on aromatase inhibitor. Oncotarget. 2016;7:74448–59. This study demonstrates the potential role of ESR1m as a biomarker for tracking disease evolution using sequencial liquid biopsies.
Fribbens C, O'Leary B, Kilburn L, et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast Cancer. J Clin Oncol. 2016;34:2961–8.
•• Schiavon G, Hrebien S, Garcia-Murillas I, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med. 2015;7:313ra182. Evaluation of the role of ESR1m as a prognostic and predicitve factor in a cohort of AI-refractory patients enrolled in the PALOMA3 trial.
Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun. 2016;7:11579.
Niu J, Andres G, Kramer K, Kundranda MN, Alvarez RH, Klimant E, et al. Incidence and clinical significance of ESR1 mutations in heavily pretreated metastatic breast cancer patients. Onco Targets Ther. 2015;8:3323–8.
Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–209.
Chu D, Paoletti C, Gersch C, VanDenBerg DA, Zabransky DJ, Cochran RL, et al. ESR1 mutations in circulating plasma tumor DNA from metastatic breast Cancer patients. Clin Cancer Res. 2016;22:993–9.
Sefrioui D, Perdrix A, Sarafan-Vasseur N, Dolfus C, Dujon A, Picquenot JM, et al. Short report: monitoring ESR1 mutations by circulating tumor DNA in aromatase inhibitor resistant metastatic breast cancer. Int J Cancer. 2015;137:2513–9.
Guttery DS, Page K, Hills A, Woodley L, Marchese SD, Rghebi B, et al. Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor-positive metastatic breast cancer. Clin Chem. 2015;61:974–82.
Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Inao T, Sueta A, Fujiwara S, et al. Clinical significance of monitoring ESR1 mutations in circulating cell-free DNA in estrogen receptor positive breast cancer patients. Oncotarget. 2016;7:32504–18.
Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Tomiguchi M, Sueta A, Murakami K, et al. Analysis of ESR1 and PIK3CA mutations in plasma cell-free DNA from ER-positive breast cancer patients. Oncotarget. 2017;8:52142–55.
Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Tomiguchi M, Sueta A, Murakami K, et al. Comparison of ESR1 mutations in tumor tissue and matched plasma samples from metastatic breast cancer patients. Transl Oncol. 2017;10:766–71.
Fribbens C, Garcia Murillas I, Beaney M, et al. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Ann Oncol. 2017;
Toy W, Weir H, Razavi P, Lawson M, Goeppert A, Mazzola A, et al. Activating ESR1 mutations differentially impact the efficacy of ER antagonists. Cancer Discov. 2017;7:277–87.
Augusto L, Sarafan-Vasseur N, Perdrix A, et al. Prognostic and predictive value of circulating ESR1 mutations in metastatic breast cancer patients (mBC) progressing under aromatase inhibitor (AI) treatment. J Clin Oncol. 2016;34
Clatot F, Perdrix A, Augusto L, Beaussire L, Delacour J, Calbrix C, et al. Kinetics, prognostic and predictive values of ESR1 circulating mutations in metastatic breast cancer patients progressing on aromatase inhibitor. Oncotarget. 2016;7:74448–59. https://doi.org/10.18632/oncotarget.12950.
Schiavon G, Hrebien S, Garcia-Murillas I, et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrated evolution during therapy for metastatic breast cancer. Sci Transl Med. 2015;7:182.
Jeselsohn RYR, Buchwalter G, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–67.
Fribbens C, O’Leary B, Kilburn L, et al. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol 2016;34:ahead of print.
Reinert T, Saad E, Barrios C, Bines J. Clinical implications of ESR1 mutations in hormone receptor-positive advanced breast cancer. Front Oncol. 2017;7:26.
Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-α: a novel mechanism for acquired endocrine resistance in breast Cancer. Cancer Res. 2013;73:6856–64.
Chandarlapaty SCD, He W, Sung P, Samoila A, You D, Bhatt T, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016;2:1310–5.
Goss PEIJ, Pritchard KI, Robert NJ, Muss H, Gralow J, Gelmon K, et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N Engl J Med. 2016;375:209–19.
Ladd B, Mazzola A, Bihani T, et al. Effective combination therapies in preclinical endocrine resistant breast cancer models harboring ER mutations. Oncotarget. 2016;7:54120–36.
Weir H, Bradbury R, Lawson M, et al. AZD9496: an oral estrogen receptor inhibitor that blocks the growth of ER-positive and ESR1-mutant breast tumors in preclinical models. Cancer Res. 2016;76:3307–18.
Mayer Iea. Phase I study of ARN-810, a novel selective estrogen receptor degrader, in postmenopausal women with locally advanced or metastatic estrogen receptor positive breast cancer [abstract]. In: CTRC-AACR san Antonio breast Cancer symposium OT3-2-07; 2013.
Joseph J, Darimont B, ZHou W. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. eLife. 2016;5. doi: https://doi.org/10.7554/eLife.15828.
Bihani T, Patel H, Arlt H. Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has anti-tumor activity in multiple ER+ breast cancer patient-derived xenograft models. Clin Cancer Res. 2017;23:4793–804.
Wardell S, Nelson E, Chao C, McDonnell J. Bazedoxifene exhibits antiestrogenic activity in animal models of tamoxifen-resistant breast cancer: implications for treatment of advanced disease. Clin Cancer Res. 2013;19:2420–31.
Wang T, et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor co-activators SRC-3 and SRC-1. Cancer Res. 2014;74:1506–17.
Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Cancer therapy: ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216–20.
Spoerke J, Gendreau S, Walter K, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun. 2016;7:11579.
Jeselson R. Are we ready to use ESR1 mutations in clinical practice? Breast Care. 2017;12:309–31.
Ng C, Schultheis A, Bidard F, Wigelt B, Reis-Filho J. Breast Cancer genomics from microarrays to massively parallel sequencing: paradigms and new insights. J Nat Cancer Inst. 2016;107
Niu J, Andres G, Kramer K, Kundranda MN, Alvarez RH, Klimant E, et al. Incidence and clinical significance of ESR1 mutations in heavily pretreated metastatic breast cancer patients. Oncotargets Ther. 2015;8:3323–8.
Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M. Droplet digital polimerase chain reaction assay for screening of ESR1 mutations in 325 breast cancer specimens. Transl Res. 2015;166:540–53.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Tomás Reinert, Rodrigo Gonçalves, and José Bines declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Breast Cancer
Rights and permissions
About this article

Cite this article
Reinert, T., Gonçalves, R. & Bines, J. Implications of ESR1 Mutations in Hormone Receptor-Positive Breast Cancer. Curr. Treat. Options in Oncol. 19, 24 (2018). https://doi.org/10.1007/s11864-018-0542-0
Published:
DOI: https://doi.org/10.1007/s11864-018-0542-0