
Abstract
Background
Helicobacter pylori (H. pylori) infection, the main cause of chronic gastritis, increases gastric cancer risk. Antibiotics-based H. pylori eradication treatment is 90 % effective. However, it is expensive and causes side effects and antibiotic resistance. Lactic acid bacteria (LAB) could present a low-cost, large-scale alternative solution to prevent or decrease H. pylori colonization.
Aim
This work aimed to study the inhibitory effects of LAB strains on the growth and pathogenic activity of H. pylori stains. To this end, we have selected the most virulent H. pylori strains (out of 20 mucosal antral biopsies) regarding cellular vacuolization and induction of apoptosis/necrosis.
Method
The selection of H. pylori pathogenic strains (clinically pre-isolated) were based on their impact of VacA activities on Hep-2 cell line, induction of apoptosis and necrosis in Caco-2 cell line. The Inhibitory effect of LAB strains on the invasion was carried out using the Caco-2 and Hela cell lines, where, they were co-cultured with the pathogenic H. pylori in the presence or absence of LAB extracts. The effect of LAB extracts on TNF-α secretion which induced by H. pylori-LPS was carried out by RT-qPCR.
Results
L. bulgaricus DSMZ 20080, L. acidophilus and L. plantarum (studied previously and reported as high antioxidant candidate strains) showed the highest anti-pylori activities with inhibition ranged from 51.46 to 88.19 %, they preventing the adhesion, invasion and DNA fragmentation of cell lines. In addition, they could reduce the TNF-α expression by 62.13 %.
Conclusion
LAB extracts could inhibit the bacterial adhesion and invasion, gastric inflammation and DNA fragmentation induced by Helicobacter pylori.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Helicobacter pylori (H. pylori) are spiral-shaped bacteria that colonize the stomach of half the populations worldwide [1]. Once a person is infected, the organism can live in the stomach indefinitely and may not cause clinical illness. It is still not clear how H. pylori are transmitted or why some of those infected become sick while others do not [2]. Although H. pylori infection is a common infection, it is not benign [1]. Close contact of H. pylori with the epithelium results in the secretion of free radicals and cytokines such as interleukin-8 IL-8, IL-1and α-TNF [3]. Excessive cytokines secretion is considered to play an important role in the alteration of epithelial proliferation, increased apoptosis oxidative DNA damages, occurrence of chronic gastritis, gastric lesions and duodenal and gastric ulcers that are the strongest known risk factors for adenocarcinoma of the distal stomach [4]. The product of particular significance for the virulent action of H. pylori is its cell wall lipopolysaccharide (LPS). The effects of H. pylori lipopolysaccharide (H. pylori-LPS) have been manifested by the marked increase of nitric oxide and proinflammatory cytokines including IL-8 in gastric mucosa [5], H. pylori-LPS is a natural ligand for Toll-like receptor 4 (TLR4) in gastric epithelia. It has been proposed that H. pylori-LPS induces IL-8 production in gastric epithelia through activating the TLR4 signaling pathway [5]. The conventional eradication therapies combine two antibiotics and a proton pump inhibitor [6]. Treatment regimens containing metronidazole, clarithromycin and a proton pump inhibitor are among the most effective [7]. The success rate following this therapy is constantly decreasing, mainly due to antibiotic resistance [8]. Resistance of H. pylori to antibiotics is a growing world concern and needs urgent public health attention [9].
Probiotics are living microorganisms with no or low pathogenicity, which exert beneficial effects on the host. They may represent a natural barrier against the development of H. pylori colonization [10]. There are several possible mechanisms by which probiotic bacteria could inhibit the adhesion of H. pylori. Certain lactobacilli exert their antiadhesion activity by secreting antimicrobial substances [11], while, strains such as L. reuteri inhibit H. pylori growth by competing with adhesion sites or by the binding of H. pylori to specific glycolipid receptors asialo-GMI and sulfatide [12]. In addition, probiotic bacteria can reduce gastrointestinal inflammation due to H. pylori-induced proinflammatory cytokines [13]. However, the signaling pathways which are modulated by LAB in gastric epithelia have not been well elucidated.
Methods
Cell lines
Caco-2 cell line (human epithelial colorectal adenocarcinoma cells), Hep-2 cell line (larynx epidermoid carcinoma) and HepG2 cell line (hepato cellular carcinoma) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM of l-glutamine, 10 % fetal calf serum (FCS) and 1 % penicillin–streptomycin solution in 50- and 100-cm2 cell culture flasks, incubated at 37 °C in a humidified atmosphere of 5 % CO2, 95 % air for 24 h.
LAB strains
Streptococcus thermophilus, Streptococcus lactis subspecies Cremoris, Lactobacillus casei, Lactobacillus delbrueckii subspecies bulgaricus, Lactobacillus delbrueckii subspecies bulgaricus DSMZ 20080 and Lactobacillus delbrueckii subspecies bulgaricus DSMZ 20081 T, Lactobacillus fermentum DSMZ 20049, Lactobacillus acidophilus DSMZ 20079 T, Lactobacillus rhamnosus, Lactobacillus plantarum subsp. plantarum DSMZ 20174 and Bifidobacterium longum subsp. Longum DSMZ 200707 were collected from culture collection of Faculty of Agriculture Ain Shams University, Faculty of Agriculture Kafr El-Sheikh University and Faculty of Science Tanta University.
Preparation of intracellular and extracellular extracts
Both extracts were prepared as previously explained by El-Adawi el al. [14]. In brief, the probiotic cultures were collected at the early stationary phase of growth and centrifuged at 10,000 rpm at 2 °C for 30 min; the culture supernatants were separated carefully and filtered through a 0.22-μm pore-size filter. For preparation of intracellular extracts, cells were washed three times with a sterile phosphate buffer solution (PBS, pH 7.4) and resuspended in sterile PBS followed by ultrasonic disruption in five 1-min intervals in an ice bath. Cell debris was removed by centrifugation at 10,000 rpm at 2 °C for 10 min, and the resulting supernatant was used as the intracellular extract, both extracellular and intracellular extracts’ stocks were stored frozen at −80 °C till used. The total cell numbers were adjusted to 109 CFU/ml for the preparation of both extracellular and intracellular extracts. The extracts stored frozen at −80 °C till used. The total cell numbers were adjusted to 109 CFU/ml for the preparation of both extracellular and intracellular extracts.
Cytotoxicity assay
For the determination of treatment concentrations that are non-toxic to Caco-2 cells, the cytotoxic assay was performed on which the non-toxic doses TC0 (recommended doses) and the concentration which kills 50 % of the cells (TC50) were determined using serial dilutions of the extracts. A cell suspension of 6 × 104 cell/ml was collected and seeded in 96-well plates (100-μl cell suspension per well). The plates were incubated at 37 °C in humidified 5 % CO2 for 24 h. After obtaining a semi confluent cell layer, the exhausted media were discarded and 100 μl of different treatment concentrations prepared in DMEM were added. Wells containing only media were used as a negative control. The cell plates were incubated at the same growth conditions for 3 days. After 3 days, 100 μl of neutral red stain was added to each well and incubated for 3 h, at 37 °C in humidified 5 % CO2 [15]. Excessive dye was discarded and the stained cells were fixed with 100-μl fixing solution (0.5 % formalin with 1 % CaCl2) for 1 min., then cells were destained in 100-μl destaining solution (50 % ethanol with 1 % glacial acetic acid) by shaking for 5 min. The stain intensity was assayed using automated ELISA microplate reader adjusted at 540 nm (reference filters 620 nm).
Isolation of H. pylori from gastric biopsy specimens
Twenty mucosal antral biopsies were collected from consecutive dyspeptic patients after obtaining their informed consent. All patients were H. pylori positive. Patients underwent routine gastrointestinal endoscopy because of abdominal complaints. Their ages ranged from 20 to 73 years. The biopsy specimens were collected in sterile tubes containing 1 ml of phosphate buffer saline solution (PBS) and were finely minced in a tissue grinder (mortar) in 1 ml of sterile PBS. One hundred microliter from each dilution was streaked onto Columbia agar plate supplemented with 10 % sheep blood, Trypticase soy agar supplemented with 10 % sheep blood and chocolate agar. The plates were incubated in 100 % humidity at 37 °C for up to 7 days in a microaerophilic gas mixture composed of 10 % CO2, 5 % O2, and 85 % N2 (Campy-Pak; Unipath S.p.A., Garbagnate Milanese, Milan, Italy). The agar plates were checked for growth from day 3 through day 7. An isolate was identified as H. pylori based on the positive catalase, oxidase, and urease reactions, typical colony morphology (small, round colonies), and the presence of characteristic-curved gram-negative bacilli on Gram-stained smears.
The selection of H. pylori virulence strains
The virulence of the infecting H. pylori strains is a major determinant of who develops disease. The two most important ones are VacA cytotoxin, which induces vacuolation of epithelial cells, and the cag pathogenicity island (cytotoxin associated gene A), which induces peptic ulceration or gastric adenocarcinoma [16]. In the current study, H. pylori virulent strains based on the VacA activity assay.
Vacuolating activity (VacA) assay
H. pylori VacA activity was evaluated in hep-2 cells by H. pylori-LPS according to Kranzer et al. [17] with some modifications. The level of vacuolization was determined by inverse microscope (magnification, 100× to 200×). H. pylori strains were considered cytotoxin positive if vacuolization was observed in 50 % of Caco-2 cells.
Assessment of apoptosis and necrosis induced by H. pylori
Assessment of apoptosis induced by Helicobacter pylori was done by an acridine orange–ethidium bromide mixture staining technique, where the determination of apoptosis was based on nuclear morphology and membrane integrity [18].
Caco-2 cells (2 × 105) were grown in 6-well plates to 70 % confluency, the cells were washed three times with prewarmed-sterilized PBS then infected with 100 μl of H. pylori suspension and plates were incubated at 5 % CO2, 37 °C for 3 h. After incubation, the plates were washed three times with prewarmed-sterilized PBS and DMEM media. After 24 h of infection at the same incubation conditions, the adherent cells were scraped with a scrapper, both floated and attached cells were obtained by centrifuged at 1,200 rpm for 10 min, resuspended in 100 μl DMEM free from antibiotics, 1 μl of stain mixture acridine orange (100 μg/ml 95 % absolute ethanol) and ethidium bromide (100 μg/ml PBS) was mixed with a 0.1-ml cell suspension. aAdrop was applied to a microscope slide and cells were then examined under a fluorescence microscope.
The efficiency of LAB in controlling Helicobacter pylori infection
Anti-Helicobacter pylori activity of LAB strains
In order to select the most potent LAB strains against H. pylori, the susceptibility of H. pylori growth to LAB extracts was evaluated by a microplate reader assay. The microplate reader (automated ELIZA microplate reader) has been adjusted at 620 nm. The inhibition percentage of LAB extracts was calculated according to the following equation.
where, A is the absorbance of the treated group, A1 is the absorbance of the blank, A0 is the absorbance of the control group.
Inhibition of H. pylori adhesion and invasion by the LAB strains
Adhesion assay
Caco-2 cells were maintained as confluent monolayers in complete DMEM medium; monolayers were washed with prewarmed PBS and trypsinized for 2 min. The cells were scraped from the flask into 72 ml of DMEM/FCS/antibiotics. Three tissue culture plates (12 wells) per flask of Caco-2 cells were prepared by depositing one sterile 13-mm glass coverslip to each well, and 2 ml of the Caco-2 cell suspension was added to each well incubated at 37 °C in the previous conditions until the monolayers were 60–70 % confluent (usually 24 h). The brucella broth medium was inoculated with H. pylori (the most potent anti-pylori strains) in the presence and absence of LAB extracts, and incubated in a microaerophilic conditions at 37 °C for 16–18 h before the assay. On the day of the assay, 2-ml fresh DMEM without supplements was added to each well, followed by 100 μl of appropriate bacterial suspension. Plates were incubated at 37 °C for 3 h. Unbound bacteria are removed by washing three times with phosphate buffered saline. Cells were washed briefly with methanol, discarded, and then mixed with fresh methanol (1 ml per well) for 1 min, which replaced after that by 2 ml per well of Giemsa stain, (freshly diluted 1:10 in water). The stain was left for 30 min at room temperature, then discarded. The monolayers were washed with 3–4 ml each of tap water and the slides were read at 100× magnification under oil immersion [19].
Invasion assay
Invasion assay was done by acridine orange–crystal violet staining technique according to Wilkinson et al. [20], with some modifications. HepG2 cells were used in this assay, both bacterial suspension and HepG2 cells were prepared as described before in the adhesion assay. Cell monolayers were infected with bacteria suspension with and without the treatments. After incubation, cells were washed in Hanks balanced salt solution stained with 0.01 % acridine orange in Gey’s solution for 45 s, rinsed with Hanks balanced salt solution, and counterstained with 0.05 % crystal violet in 0.15 N NaCl for 45 s. After the coverslips were rinsed with Hanks balanced salt solution, they were mounted on the slides. Slides were then viewed under a fluorescence microscope using incident light at 40× magnification for screening. All infecting bacteria took up the acridine orange and fluoresced; crystal violet quenched the fluorescence of extracellular adhering bacteria, so that, only fluorescent intracellular bacteria would be visible under fluorescent light microscopy. The acridine orange–crystal violet staining technique revealed viable green-fluorescing intracellular organisms. Nonviable intracellular bacteria would have stained red. The change in color is attributed to the increase in the amount of acridine orange interchelating with the phosphate–sugar backbone of DNA as the DNA becomes denatured in nonviable cells.
DNA fragmentation assay
A distinctive feature of apoptosis at the biochemical level is DNA fragmentation. This method was used as a semi-quantitative method for measuring apoptosis. Such phenomenon, described for the first time by Wyllie [21], can be visualized by an agarose gel electrophoresis analysis. The present protocol provides a method for qualitative determination of DNA fragmentation induced by H. pylori in the presence and absence of treatment (LAB).
Anti-inflammatory activities of LAB by detection of TNF- α production
Isolation of Helicobacter pylori lipopolysaccharide
In order to isolate H. pylori-LPS which plays an essential role in free radicals and immune response induction, a modified method by Uchida and Mizushima [22] was used. Bacteria were cultured in microaerophilic at 37 °C for 24 h in brucella broth supplemented with 10 % fetal bovine serum. After incubation, cells were collected by centrifugation at 10,000 rpm and 2 °C for 30 min, then washed with sterilized PBS and lyophilized. LPS was extracted from the lyophilized cells in three steps:
-
(a)
The recovery of LPS in insoluble form together with some denatured proteins
The lyophilized bacterial cells (100 mg as dried matter) were mixed with 2.2 ml of distilled water. To the mixture was added successively 0.4 ml of 100 mM Tris–HCl (pH 8.0), 0.4 ml of 0.5 M MgCl2 and 1 ml of 8 % triton X-100. The tube, tightly capped, was heated in boiling water bath for 10 min, after cooling, the mixture was centrifuged at 10,000 rpm for 15 min and then the precipitate was washed once with 4 ml of 10 mM Tris–HCl (pH 8.0) and 10 mM MgCl2.
-
(b)
The solubilization of LPS
To the obtained precipitate, 1 ml of distilled water, 0.2 M EDTA (pH 8.0), 2 M NaCl and 8 % Triton-X 100 were added, and the suspension was mixed well. The mixture was incubated at 37 °C for 60 min with shaking and then centrifuged at 10,000 rpm for 15 min. The supernatant was then collected in another centrifuge tube.
-
(c)
Precipitation of LPS
The collected supernatant (3.6–4 ml) was added 0.4 ml of 1 M MgCl2 under stirring and then the mixture was incubated at 37 °C for 60 min. The resulted opaque solution was immediately centrifuged at 10,000 rpm for 90 min at 15 °C and the transparent precipitate that formed was further treated with proteinase K (100 μg/ml) at 37 °Cfor 2 h before the final washing with 4 ml of 10 mM Tris–HCL (pH 8.0) and 10 mM MgCl2.
The preparation was further treated with proteinase K (20 μg/ml) at 37 °C for 2 h before the final washing.
PBMC cells (2 × 105) were grown in RPMI medium and seeded into 96-well plates and incubated for 24 h prior to stimulation with H. pylori-LPS (10 mg/ml). That stimulation was carried out in the presence and absence of LAB extracts and left for 24 h.
RNA extraction and real-time RT-PCR analyses
Extraction of total cells RNA was performed with guanidine isothiocyanate according to the method by Chomczynski and Sacchi [23]. First-strand cDNA was synthesized using oligo-dT primer and the AMV reverse transcriptase (Promega Corp., Madison, WI, USA). In brief, 5 μl of total RNA was reversely transcribed by adding 20 ml of a master mixed with 1 U/ml RNase inhibitor, 5 mmol/l MgCl2, 1× reverse transcriptase buffer, 1 mmol/l of dNTP mixture, and 0.7 U/ml AMV reverse transcriptase (final concentrations indicated). Samples were incubated at 42 °C for 60 min. For the cDNA amplification, the PCR with hot start technique was used, in which Taq polymerase was added to each tube with Thermal Cycler. The PCR mixture contained a final concentration of 10 pmol/l specific primers for TNF-α, 1× PCR buffer, 1.5 mmol/l MgCl2, 0.2 mol/l of each dNTP and 1 U/25 ml Taq polymerase. After a 5-min initial melting step at 97 °C, the PCR with 28–32 cycles was carried out [94 °C, 1 min for denaturation; 58 °C (TNF-a), 1 min for annealing; and 72 °C, 1 min for extension]. The final cycle was followed by a 10-min soak at 72 °C. The sequences of primer pairs and predicted sizes of the amplified PCR fragments were shown in Table 1. The house keeping gene GAPDH was used as an internal control for the standardization of the PCR product. The RT-PCR was done to cDNA based on the SYBR Green I dye and LightCycler fluorimeter. The RT step was followed by first round PCR using the TNF-α primers in a 50-μl reaction containing 3–5 μl cDNA, 10 pmol from each of the forward and reverse primers and 12.5 μl master mix SYBR Green. The thermal cycling protocol was as follows: 30 cycles of 1 min each at 94, 56 and 72 °C, with the final extension done at 72 °C for 5 min.
Primers
Primers | Sequence |
|---|---|
TNF-α forward | 5′-TTC TGT CTA CTG AAC TTC GGG GTG ATG GGT CC-3′ |
TNF-a reverse | 3′-GTA TGA GAT AGC AAA TCG GCT GAC GGT GTG GG-5′ |
GAPDH forward | 5′-GAA GGT GAA GGT CGG AGT |
GAPDH reverse | 3′-GAA GAT GGT GAT GGG ATT TC |
Results
Cytotoxicity assay of LAB extracellular extract on Caco-2 cell line and peripheral blood mononuclear cell (PBMC)
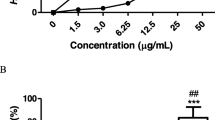
The cytotoxicity results indicated that intracellular extracts were safer than extracellular extracts on Caco-2 cells. In general, the treatments have no TC50 even in the maximum concentrations. The safe doses of extra- and intra-cellular extracts on Caco-2 cells ranged from 4 to 14 and 6 to 18 % respectively (Table 1).
Isolation of H. pylori from gastric biopsy specimens
The clinical history of H. pylori infected patients revealed that generally there is a positive correlation between H. pylori infection and liver fibrosis diseases (data sheet). The variations in symptoms started from abdominal pain and gastritis to ulcers, with aging this converted to malignant ulcers and multilymphoma. The endoscopic photos of patients (Fig. 1) showed antral gastritis, malignant ulcer and incisural mass in comparison to normal pylorus and antrum of non-infected people.

Endoscopic photos of patients undergoing endoscopy
The selection of H. pylori virulence strains
The virulent H. pylori strain should have the ability to induce vacuolation and apoptosis or necrosis in cultured epithelial cell lines. Vacuolating activity results indicated that the ability of the isolates to induce vacuoles in cell lines ranged from weak and moderate to high (Fig. 2). The isolates no. 3, 4, 5, and 6 recorded the highest vacuolating activity (Table 2). In addition to the vacuolating activity, all isolates had the ability to induce all stages (early to late) of apoptosis (Fig. 3) and necrosis (Fig. 4) by converting the nucleus into multinucleated cells, condensed nucleated cells and chromatin fragmented cells that were visualized with acridine orange–ethidium bromide fluorescent stains.

Vaculating activity of the H. pylori clinical isolates on Hep-2 cells

Assessment of apoptosis and necrosis induced by Helicobacter pylori. Apoptotic cells had condensed and/or fragmented nuclei, these cells were impermeable to ethidium bromide during early stages and their nuclei stained green but during late stages, this ability was lasted and their nuclei stained red. Necrosis cells: cells exhibited a red nucleus stain but with no nuclear condensation they exhibited either normal nuclear structure or had no nuclear staining

Inhibition of H. pylori–Hep-2 adhesion by lactic acid bacteria. a, b Hep-2 mammalian cells infected with H. pylori. c Hep-2 mammalian cells infected with H. pylori treated with LAB. d Hep-2 mammalian cells (at high magnification) infected with H. pylori treated with LAB. e Hep-2 mammalian cells (control). The small dark arrows in a, b pointed to H. pylori adhered to cell in the spiral shape, while in c, d pointed to the H. pylori under lactic acid bacterial treatment converted to coccid forms with reduction in their ability to adhered to cells
The efficiency of LAB in controlling H. pylori
The antibacterial activity of LAB extracts indicated that, in general, the extracellular extracts of the LAB were more effective than the intracellular extract. The extracellular bacterial extracts of L. plantarum, L. fermentum, L. bulgaricus DSMZ 20080 and S. lactis were the most potent antibacterial treatments (Table 3). The antibacterial activity of these treatments ranged from 81.44 to 88.19 %. In addition, three H. pylori strains’ isolates (no. 2, 3 and 4) were the most resistant strains to the treatment, while strain no. 5 was the most sensitive strain to the treatments followed by strain no. 6 then no. 1.
Inhibition of H. pylori–Hep-2 adhesion by LAB
The pretreatment of H. pylori with the most potent antibacterial extracts prevents the remaining viable H. pylori from adhering to cell lines. In addition, after treatments, they conversed morphologically into a coccoid form either was viable but non-culturable or was killed, in comparison to the control group (in which the non-treated helical H. pylori were able to adhere to the cell lines). Visual inspection of Hep-2 cells after the infection/replication phases demonstrated internalized bacteria, sometimes enclosed in apparent vacuoles (Fig. 5). The outer integument of Hep-2 cells had often lost its integrity and the cytosol was distorted and morphologically difficult to observe.

Inhibition of H. pylori invasion to HepG2 cells by LAB; a the control HepG2 cells, b Hepg2 cells treated with LAB and infected with H. pylori, c H. pylori
Inhibition of H. pylori invasion to HepG2 cells by LAB
HepG2 cells infected with H. pylori, green fluorescent bacteria, could be detected within HepG2 cells (indicating invasion) after staining with acridine orange (Fig. 6). The pretreatment of H. pylori with the most potent antibacterial extracts decreases the number of the invading bacteria from 20 cells to single H. pylori cell.

Inhibitory effect of LAB on DNA fragmentation induced by H. pylori L ladder DNA. C DNA of the non-infected cell (control). I DNA of cells infected cells with H. pylori. T DNA of cells infected with H. pylori previously treated with LAB
Inhibition of DNA fragmentation induced by H. pylori by LAB
During apoptosis a series of reorganisation cascades occur in the cell such as DNA fragmentation, which is a biochemical hallmark of apoptosis in the majority of cells. An endogenous Ca++- and Mg++-dependent endonuclease is believed to be responsible for DNA cleavage by breaking the double strand DNA at inter-nucleosomal sites. As shown in figure, the DNA of mammalian cell infected with H. pylori showed apoptotic DNA cleavage (characteristic oligonucleosomal fragments-lane I) whereas, in case of with the most potent antibacterial LAB treatment, DNA mammalian cell appears as non-segmented bands when compared with the infected one.
Anti-inflammatory activities of LAB
Reduction of TNF-α production
qRT-PCR results clarified that the most potent LAB extracts (co-treatment model) could reduce TNF-α secretion in LPS-stimulated model (Fig. 7). Referring to the GAPDH house keeping gene, the LAB treatment reduced the TNF-α section with a percentage of 62.13 (Table 4).

Anti-inflammatory activities of LAB. L ladder DNA. C PCR-amplified gene fragment of control cell (non-treated with LAB). T PCR-amplified gene fragment of cells treated with LAB
Discussion
Cytotoxicity assay was done (Table 1) to determine the non-toxic doses TC0 (recommended doses) and the concentration which kills 50 % of the cells (TC50), using serial dilutions of the extracts; the safe doses should have a maximum inhibition percentage that does not exceed 10 %, i.e. the percentage of surviving cells is not less than 90 %. Neutral red (NR) is a weak cationic supravital dye that readily penetrates cell membranes by non-ionic diffusion and predominately accumulates intracellularly in lysosomes. Alterations of the cell surface or the sensitive lysosomal membrane lead to lysosomal fragility and other changes that gradually become irreversible. Thus, the uptake and binding of NR decrease in altered cell membrane integrity making it possible to distinguish between viable, damaged or dead cells via spectrophotometric measurements.
The clinical history of H. Pylori-infected patients
Recently, evidence showing a possible association between H. pylori infection in the human stomach and chronic liver diseases has emerged [24]. Moreover, the prevalence of H. pylori in patients with cirrhosis has been reported to be remarkably higher than in non-cirrhotic patients [25], these observations support the idea that H. pylori infection may have a critical role in the pathogenesis of various liver disorders. This was confirmed in the current study where the positive correlation between H. pylori infection and various liver disorders was revealed (Fig. 1).
Data sheet of the clinical history of consecutive dyspeptic patients undergoing endoscopy
No. | Age | Gender | Smoking | Diabetes mellitus | Cholesterol level | Coronary heart disease | Liver diseases (fibrosis) | Endoscopic picture | Anemia |
|---|---|---|---|---|---|---|---|---|---|
1 | 73 | Male | + | + | − | − | Post portal fibrosis | Gastritis | + |
2 | 60 | Female | − | − | − | − | Cirrhosis | Malignant ulcer, multilymphoma | + |
3 | 65 | Male | − | − | − | − | − | Ulcer | + |
4 | 50 | Female | − | − | + | − | − | Gastric cancer | + |
5 | 70 | Male | + | − | − | − | Cirrhosis | Gastritis | + |
6 | 60 | Female | − | − | − | − | Cirrhosis | Malignant ulcer | + |
7 | 27 | Female | − | − | − | − | − | Gastritis | + |
8 | 20 | Female | − | − | − | − | − | Ulcer | + |
9 | 27 | Female | − | − | − | − | − | Malignant ulcer | − |
10 | 66 | Male | − | − | − | − | Post portal fibrosis | Gastric cancer | + |
11 | 33 | Male | + | − | + | − | − | Gastritis | − |
12 | 55 | Female | − | − | − | − | Cirrhosis | Malignant ulcer | + |
13 | 50 | Male | + | − | − | − | Post portal fibrosis | Gastritis | + |
14 | 60 | Male | + | − | + | − | Post portal fibrosis | Malignant ulcer | + |
15 | 69 | Male | + | − | − | − | Cirrhosis | Gastritis | + |
16 | 42 | Male | + | − | − | − | − | Gastritis | + |
17 | 30 | Female | − | − | − | − | − | Gastritis | + |
18 | 20 | Female | − | − | − | − | − | Gastritis | − |
19 | 66 | Male | − | − | − | − | Cirrhosis | Malignant ulcer | + |
20 | 50 | Male | + | − | − | − | Cirrhosis | Gastric cancer | + |
The virulence factor of H. Pylori
The VacA toxin is a major virulence factor of Helicobacter pylori [26]. In H. pylori growth medium, it forms trans-membrane anion-specific channels [27], which are likely to contribute to vacuolization by causing osmotic swelling of such acidic compartments where the toxin localizes following endocytosis [28]. VacA can also induce vacuoles from the cytosol upon transfection [29]. In the present work, we selected four H. pylori isolates out of six having high ability to induce vacuoles as observed in results section (Table 2; Fig. 2). The enhanced gastric epithelial cell apoptosis in H. pylori infection has been suggested to play an important role in the pathogenesis of chronic gastritis, peptic ulcer and gastric neoplasia. There are a number of mechanisms have been postulated by Wu et al. [30], including the direct cytotoxic effects of the bacteria, as well as inflammatory responses elicited by the infection. Wu et al. [30] suggested that T helper type 1 (Th1) cells are selectively increased during H. pylori infection. Th1 cytokines, such as gamma interferon (IFN-) and tumor necrosis factor alpha (TNF-α), can increase the release of proinflammatory cytokines, augmenting apoptosis induced by H. pylori. In the present work, the four isolates have the high ability to induce all stages of apoptosis (Fig. 3) as well as necrosis (Fig. 4).
The efficiency of LAB in controlling H. pylori pathogenicity
Inhibition of H. pylori growth
LAB has been reported to inhibit the adhesion of many kinds of pathogenic enterobacteria to intestinal cells [31]. In the present study, the LAB extracellular extract could inhibit H. pylori by more than 88 % (Table 3). This inhibition could be related to the bacteriocin family as reported by Cats et al., 2003 [32]. Other known substances secreted by these bacteria are the endproducts of lactic acid fermentation, such as lactic and acetic acids, and hydrogen peroxide [33].The production of relatively large amounts of lactate by lactobacilli has been implicated as an inhibitory factor of H. pylori by some authors [34]. Lactic acid, in addition to its antimicrobial effect resulting from the lowering of the pH, could inhibit the H. pylori urease. However, the inhibitory effects of lactobacilli on H. pylori differ from strain to strain as reported by [35]. In the current study, L. plantarum, L. fermentum, L. bulgaricus DSMZ 20080 and S. lactis were the most potent antibacterial out of eleven strains. In conclusion, the in vitro inhibitory effect of certain LAB is probably related to lactic acid and/or other antimicrobial substances yet to be identified.
Inhibition of H. pylori–Hep-2 adhesion by LAB
The adhesion of H. pylori to epithelial cells is important in determining the outcome in H. pylori-associated diseases [36]. In the gastric mucosa, H. pylori possibly interact with epithelial cells through secretory components or as a result of adherence [37]. Some studies showed that L. johnsonii, L. salivarius and L. acidophilus inhibit the attachment of H. pylori to intestinal HT-29 cells [38] or to MKN 45 gastric cell lines [39]. The current study confirmed the previous studies where LAB could inhibit the adhesion of H. pylori to the host cells Hep-G2 (Fig. 5). There are several possible mechanisms by which LAB can inhibit the adhesion of H. pylori. The first mechanism was postulated by Mukai et al. [12] on which the competition mechanism has been assumed. For example, it has been demonstrated that L. reuteri inhibited the binding of H. pylori to specific glycolipid receptors asialo-GMI and sulfatide. However, a nonspecific rather than a specific blockage of receptor sites is the most likely mechanism because lactobacilli can inhibit adhesion of large varieties of pathogenic bacteria, although each adheres to its particular receptor on the cells. The second mechanism might be via an on/off switching of BabA (adherence factor blood group Ag-binding adhesion) gene expression of H. pylori which facilitates bacterial colonization and augments a nonspecific immune response 1. The third mechanism we suggested here is the protective effect of LAB on the integrity of the host cells which prevent the adhesion of H. pylori.
Inhibition of H. pylori–Hep-2 invasion by LAB
There are two classical virulence determinants expressed by H. pylori: the Cag A protein encoded by the cytotoxin-associated genes pathogenicity island (cagPA1) and the vacuolating cytotoxin (VacA). In this section, we discuss the inhibition of H. pylori–Hep2 invasion by LAB (Fig. 6). The inhibition mechanism was postulated by Ryan et al. [40], who found that CagA accumulated in H. pylori cells following exposure to L. salivarius presumably as a result of loss of functionality of the Cag secretion system. These data identified a new mechanism whereby some LAB have a positive effect on H. pylori-associated inflammation without clearing the infection.
Inhibition of DNA fragmentation
The molecular mechanisms of H. pylori-associated gastric carcinogenesis remain ill defined. In this study, we examined the possibility that H. pylori directly compromise the genomic integrity of its host cells. We provide evidence that the infection introduces DNA double-strand breaks (DSBs). The induction of DSBs depends on the direct contact of live bacteria with mammalian cells. The infection-associated DNA damage is evident upon separation of nuclear DNA (Fig. 7). Bacterial adhesion (e.g., via blood group antigen-binding adhesin) is required to induce DSBs. In contrast, the H. pylori virulence factors vacuolating cytotoxin A, γ-glutamyl transpeptidase and the cytotoxin-associated gene (Cag) pathogenicity island are dispensable for DSB induction [41]. Although most breaks are repaired efficiently upon termination of the infection, the prolonged active infection might lead to saturation of cellular repair capabilities. In summary, we conclude that DNA damage followed by potentially imprecise repair is consistent with the carcinogenic properties of H. pylori and with its mutagenic properties in vitro, in vivo it may contribute to the genetic instability and frequent chromosomal aberrations that are a hallmark of gastric cancer.
The anti-inflammatory effect of LAB
The inflammatory response to gastric H. pylori infection is characterized by the release of various inflammatory mediators such as chemokines and cytokines. A large body of evidence has shown that H. pylori-LPS-induced inflammation in gastric mucosa has nearly the same pathological characteristics as the mucosal inflammation initiated by H. pylori infection [5]. Bhattacharyya et al. [42] suggested that H. pylori-LPS may be a major virulent factor for H. pylori-associated mucosal inflammation, which urged us to research the effect of Lactobacilli on H. pylori-LPS-induced cytokines production. The cytokine response is initially manifested by the release of interleukin 8 (IL-8), which leads to the migration of neutrophils and monocytes to the mucosa [43]. Activated monocytes and dendritic cells in the lamina propria produce tumor necrosis factor (TNF-α) as shown on Fig. 7-lane C, as well as IL-1 and IL-6 [42], IL-1 and IL-6 stimulate CD41 T cells (type 1), and these produce a variety of cytokines including IL-4, IL-5, IL-6, and interferon-g [44]. This response is unable to clear the infection and sustains inflammation. LAB could modify the immunologic response of the host by interacting with epithelial cells and modulating the secretion of anti-inflammatory cytokines, which would result in a reduction of gastric activity and inflammation [45], shown on Fig. 7—lane T. However, the effect of LAB on the immune response is difficult to generalize. Distinct LAB strains may generate divergent immune responses, which, in turn, depend on the host’s immune status [46].
References
Stege PW, Davicino RC, Veja AE, Casali YA, Correa S, Micalizzi B (2006) Antimicrobial activity of aqueous extracts of Larrea divaricata Cav (jarilla) against Helicobacter pylori. Phytomedicine 13:724–727
Baldari CT, Lanzavecchia A, Telford JL (2005) Immune subversion by Helicobacter pylori. Trends Immunol 26:199–207
Noach LA, Bosma NB, Jansen J, Hoek FJ, van Deventer SJ, Tytgat GN (1994) Mucosal tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8 production in patients with Helicobacter pylori infection. Scand J Gastroenterol 29:425–429
Peek RM, Crabtree JE (2006) Helicobacter infection and gastric neoplasia. J Pathol 208:233–248
Ogawa T, Asai Y, Sakai Y et al (2003) Endotoxic and immunobiological activities of a chemically synthesized lipid A of Helicobacter pylori strain 206-1. FEMS Immunol Med Microbiol 36:1–7
Castillo-Juárez I, González V, Jaime-Aguilar H et al (2009) Anti-Helicobacter pylori activity of plants used in Mexican traditional medicine for gastrointestinal disorders. J Ethnopharmacol 122(2):402–405
De Francesco V, Zullo A, Ierardi E et al (2009) Phenotypic and genotypic Helicobacter pylori clarithromycin resistance and therapeutic outcome: benefits and limits. J Antimicrob Chemother 65:327–332
Wolle K, Malfertheiner P (2007) Treatment of Helicobacter pylori. J Clin Gastroenterol 21(2):315–324
Jenks PJ, Edwards D (2002) Metronidazole resistance in Helicobacter pylori. Int J Antimicrob Agents 19:1–7
Weizman Z, Asli G, Alsheikh A (2005) Effect of a probiotic infant formula on infections in child care centers: comparison of two probiotic agents. Pediatrics 115:5–9
Michetti P, Dorta G, Wiesel PH et al (1999) Effect of whey-based culture supernatant of Lactobacillus acidophilus (johnsonii) La1 on Helicobacter pylori infection in humans. Digestion 60:203–209
Mukai T, Asasaka T, Sato E, Mori K, Matsumoto M, Ohori H (2002) Inhibition of binding of Helicobacter pylori to the glycolipid receptors by probiotic Lactobacillus reuteri. FEMS Immunol Med Microbiol 32:105–110
Linsalata M, Russo F, Berloco P et al (2004) The influence of Lactobacillus brevis on ornithine decarboxylase activity and polyamine profiles in Helicobacter pylori-infected gastric mucosa. Helicobacter 9:165–172
El-Adawi H, Khalil AM, El-Sheekh MM, El-Deeb NM, Hussein MZ (2012) Cytotoxicity assay and antioxidant activities of the lactic acid bacterial strains. Afr J Microbiol Res 6(8):1700–1712
Borenfreund E, Puerner JA (1985) Toxicity determined in vitro by morphological alterations and neutral red absorption. J Toxicol Lett 24:119–124
Backert S, Clyne M (2011) Pathogenesis of Helicobacter pylori infection. Helicobacter 16(S1):19–25
Kranzer K, Söllner L, Aigner M, Schneider-Brachert W (2005) Impact of Helicobacter pylori virulence factors and compounds on activation and maturation of human dendritic cells. Infect Immun 73(7):4180–4189
Rena J, Peter J, Nicola L (2004) Helicobacter pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway. Infect Immun 72(5):2889–2898
Knutton S, Lloyd DR, McNeish AS (1987) Adhesion of enteropathogenic Escherichia coli to human intestinal enterocytes and cultured human intestinal mucosa. Infect Immun 55(1):69–77
Wilkinson SM, Uhl JR, Kline BC, Cockerill FR 3rd (1998) Assessment of invasion frequencies of cultured HEp-2 cells by clinical isolates of Helicobacter pylori using an acridine orange assay. J Clin Pathol 51:127–133
Wyllie AH (1980) Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284(5756):555–556
Uchida K, Mizushima S (1987) A simple methods for isolation of lipopolysaccharide from Pseudomonas aeruginosa and some other bacterial strains. Agric Biol Chem 51:3107–3114
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-chloroform extraction. Anal Biochem 162:156–159
Stalke P, Abu Al-Soud W, Bielawski KP et al (2005) Detection of Helicobacter species in liver and stomach tissues of patients with chronic liver disease using polymerase chain reaction—denaturing gradient gel electrophoresis and immunohistochemistry. Scand J Gastroenterol 40:1032–1041
Calvet X, Navarro M, Gil M et al (1997) Seroprevalence and epidemiology of Helicobacter pylori infection in patients with cirrhosis. J Hepatol 26(6):1249–1254
Parsonnet J (1998) Helicobacter pylori: the size of the problem. Gut 43(Suppl 1):S6–S9
Tombola F, Carlesso C, Szabò I et al (1999) Helicobacter pylori vacuolating toxin forms anion-selective channels in planar lipid bilayers: possible implications for the mechanism of cellular vacuolation. Biophys J 76(3):1401–1409
Montecucco C, Papini E, de Bernard M et al (1999) Molecular and cellular activities of Helicobacter pylori pathogenic factors. FEBS Lett 452(1–2):16–21
de Bernard M, Burroni D, Papini E et al (1998) Identification of the Helicobacter pylori VacA toxin domain active in the cell cytosol. Infect Immun 66(12):6014–6016
Wu Y-Y, Tsai H-F, Lin W-C et al (2004) Helicobacter pylori enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in human gastric epithelial cells. World J Gastroenterol 10(16):2334–2339
Bernet MF, Brassart D, Neeser JR, Servin AL (1994) Lactobacillus acidophilus LA 1 binds to cultured human intestinal cell lines and inhibits cell attachment and cell invasion by enterovirulent bacteria. Gut 35(4):483–489
Cats A, Kuipers EJ, Bosschaert MAR et al (2003) Effect of frequent consumption of a Lactobacillus casei-containing milk drink in Helicobacter pylori-colonized subjects. Aliment Pharmacol Ther 17:429–435
Vandenbergh PA (1993) Lactic acid bacteria, their metabolic products and interference with microbial growth. FEMS Microbiol Rev 12:221–238
Sgouras D, Maragkoudakis P, Petraki K et al (2004) In vitro and in vivo inhibition of Helicobacter pylori by Lactobacillus casei strain Shirota. Appl Environ Microbiol 70(1):518–526
Guruge JL, Falk PG, Lorenz RG, Dans M et al (1998) Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc Natl Acad Sci USA 95:3925–3930
Smoot DT (1997) How does Helicobacter pylori cause mucosal damage? Direct mechanisms. Gastroenterology 113(6 Suppl):S31–S34
Coconnier MH, Lievin V, Bernet-Camard MF, Hudault S, Servin AL (1997) Antibacterial effect of the adhering human Lactobacillus acidophilus strain LB. Antimicrob Agents Chemother 41(5):1046–1052
Nam HHM, Bae O, Lee Y (2002) Effect of Weissella confusa strain PL9001 on the adherence and growth of Helicobacter pylori. Appl Environ Microbiol 68:4642–4645
Jack RW, Tagg JR, Ray B (1995) Bacteriocins of gram-positive bacteria. Microbiol Mol Biol Rev 59(2):171–200
Ryan KA, Daly P, Li Y, Hooton C, O’Toole PW (2008) Strain-specific inhibition of Helicobacter pylori by Lactobacillus salivarius and other lactobacilli. J Antimicrob Chemother 61(4):831–834
Tegtmeyer N, Wessler S, Backert S (2011) Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J 278:1190–1202
Bhattacharyya A, Pathak S, Datta S, Chattopadhyay S, Basu J, Kundu M (2002) Mitogen-activated protein kinases and nuclear factor-kappaB regulate Helicobacter pylori-mediated interleukin-8 release from macrophages. Biochem J 368:121–129
Arakawa T, Higuchi K, Fujiwara Y et al (2000) Helicobacter pylori: criminal or innocent bystander? J Gastroenterol 35(Suppl 12):42–46
Harris RA, Owens DK, Witherell H, Parsonnet J (1999) Helicobacter pylori and gastric cancer: what are the benefits of screening only for the CagA phenotype of H. pylori? Helicobacter 4:69–76
Gill HS, Rutherfurd KJ, Cross ML, Gopal PK (2001) Enhancement of immunity in the elderly by dietary supplementation with the probiotic B lactis HN019. Am J Clin Nutr 74:833–839
Haller D, Bode C, Hammes WP, Pfeifer AM, Schiffrin EJ, Blum S (2000) Non-pathogenic bacteria elicit a differential cytokine response by intestinal epithelial cell/leucocyte co-cultures. Gut 47:79–87
Acknowledgments
We thank Dr. Hanan Sloiman and Dr. Asem Elfert (Department of Tropical Medicine & Infectious Diseases, Faculty of Medicine-Tanta University) for kindly providing the biopsy specimens.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
El-Adawi, H., El-Sheekh, M., Khalil, M. et al. Lactic acid bacterial extracts as anti-Helicobacter pylori: a molecular approach. Ir J Med Sci 182, 439–452 (2013). https://doi.org/10.1007/s11845-013-0909-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11845-013-0909-y