
Abstract
Molybdenite (MoS2) is the most commonly used mineral for molybdenum extraction. In this work, molybdenum (Mo) powder was directly synthesized via CaO-assisted carbothermic reduction of MoS2. The experimental results showed that MoS2 first reacted with CaO to form CaMoO4, Mo and CaS, and then carbon black reduced the CaMoO4 to yield Mo. Almost all the sulfur as MoS2 was fixed in CaS, and the main gaseous product was CO. In addition, it was observed that MoS2 can be almost fully converted to Mo at a MoS2:CaO:C molar ratio of 1:4:1.7 after reacting at 1200°C for 60 min. After removing by-product CaS, the prepared Mo powder has a carbon content of 0.18% and a sulfur content of 0.46%.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
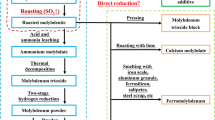
It is well known that molybdenite is the most commonly used mineral for extracting molybdenum.1 The industrial route for extracting molybdenum from molybdenite consists of multiple steps, such as oxidation roasting of molybdenite, chemical purification of the industrial molybdenum oxide, and the reduction of MoO3 by H2 to fabricate molybdenum metal powder.2,3 Even if this industrial route has been well established, it has two shortcomings. First, due to the numerous steps involved, the resource utilization rate of Mo will be decreased. In the step of the oxidation roasting of molybdenite, the recovery rate of molybdenum is about 98.5%. In the step of chemical purification of the industrial molybdenum oxide by ammonia leaching, the recovery rate of molybdenum is about 94%. However, the loss of molybdenum in the step of hydrogen reduction of MoO3 to prepare molybdenum powder can be negligible. Thus, the overall recovery rate of Mo is about 92.6%. Secondly, the step of oxidation roasting of molybdenite emitting a lot of SO2 is a very polluting process. To shorten the extraction process and to reduce environmental pollution, many strategies for directly treating molybdenite have been proposed, such as pressure oxidative leaching,4,5 lime roasting,6 chlorination roasting,7 sodium chlorate extraction,8 vacuum decomposition,9,10,11 and acidic leaching.12 In the 1970s, the strategy of the direct reduction of sulfide minerals (molybdenite, cuprous sulfide, cupric sulfide, etc.) with a desulfurizer was first proposed by Habashi and Dugdale.13 They reported that CaO as the desulfurizer can greatly facilitate the reduction of molybdenite by H2. However, after a reaction at 800°C for 1 h, the desulfurization rate was only about 40% in the presence of a stoichiometric amount of CaO. Since then, many scholars have conducted detailed studies on the lime-assisted reduction of MoS2 with H2,14,15,16,17 CO,14,18,19 C,20,21 and CH4.22,23 Rao and Prasad18 prepared molybdenum carbide (Mo2C) by reducing molybdenite with CO in the presence of CaO, and their results demonstrated that CaO can improve the conversion rate of MoS2 to CaS by over 20 times relative to that without CaO. Moreover, Rao and Prasad14 have published detailed information on the lime-scavenged reduction of MoS2 with H2, CO, and C. It has been reported that H2 is a better reducing agent because the purity of the product achieved by H2 reduction is higher than that acquired by carbothermal reduction. However, it has also been reported that sulfur-containing gas is generated when the reducing agent is H2 or CO. Therefore, C seems to be an ideal reducing agent from the perspective of a sulfur-free gaseous product. To explore the mechanism of CaO-assisted C reduction of MoS2, both Padilla et al. 20 and Tripathy et al.21 have studied the reduction of molybdenite with carbon (activated carbon or graphite) in the presence of CaO. However, the reaction mechanism has not been fully revealed. Additionally, it has been reported that the formation of Mo powder is inevitably accompanied by the formation of Mo2C.
Many studies13,14,15,16,17,18,19,20,21,22,23 have shown that lime has the potential to be an effective desulfurizer for molybdenum extraction from molybdenite. In addition, among the reductants of H2, CO, CH4, and C, since the reduction of MoS2 by CO or CH4 will result in the formation of Mo2C,14,18,19,22,23 H2 and C can be considered as the promising reducing agents for the synthesis of Mo from MoS2. Moreover, compared to H2, C has obvious advantages because of its low cost, good safety, and easy operation. However, there are still some key issues that need to be resolved for CaO-assisted carbothermic reduction of MoS2 to product Mo powder.
This work aims to explore the appropriate conditions to prepare Mo rather than the mixture of Mo and Mo2C. Here, the CaO-assisted reduction of MoS2 by carbon black was studied in detail by changing the MoS2:CaO:C molar ratio in order to illustrate the reaction mechanism and determine the optimal conditions. By adjusting the addition amounts of CaO and C to avoid the generation of Mo2C, Mo powder can be directly synthesized via CaO-assisted carbothermic reduction of MoS2 at a MoS2:CaO:C molar ratio of 1:4:1.7 after reacting at 1200°C for 1 h. More importantly, the reaction mechanisms in the MoS2-CaO binary system and the MoS2-CaO-C ternary system were studied in detail.
Experimental Procedures
Materials
Molybdenum disulfide (MoS2, 98.5 wt.% purity; Sinopharm Chemical Reagent, China) was used as the molybdenum source. CaO powder was prepared by roasting calcium carbonate (CaCO3, 99 wt.% purity; Sinopharm Chemical Reagent) at 1000°C for 10 h. Carbon black (C, 99 wt.% purity; Mitsubishi Chemical, Japan) was used as the reducing agent. The micrographs of the raw materials, MoS2 and CaO, are shown in Fig. S1. MoS2 has a typical two-dimensional structure with a lateral size of several microns, while CaO particles are composed of small grains with the size of about 500 nm.
Synthesis of Mo Powder
The isothermal experiments were conducted in a horizontal resistance furnace under argon flow. In each experiment, an approximately 5 g sample was placed in an alumina crucible. Subsequently, the crucible was placed into a quartz tube and argon gas (100 ml/min) was introduced to exclude air. When the furnace was heated to a desired temperature (800°C, 1000°C, or 1200°C), the quartz tube was placed inside and maintained at this temperature for a period of time. After the reaction was completed, the quartz tube was taken out to cool the sample to ambient temperature. Thereafter, the sample was leached in a hydrochloric acid solution. Finally, the leached residue was washed several times with deionized water and dried for subsequent testing. A schematic of the synthesis process of Mo powder is shown in Fig. 1.

(a) Schematic of the experimental apparatus. (b) Flow diagram of the synthesis process of Mo powder.
Characterization
The phase composition of the prepared specimens was evaluated by x-ray diffraction [XRD; TTR III; Rigaku, Japan; x-ray wavelength, 1.5418 Å (Cu-Kα)]. The morphology of the prepared samples was characterized by a field emission scanning electron microscope (FE-SEM; ZEISS SUPRA 55; Oberkochen, Germany) and a transmission electron microscope (TEM; Tecnai-G2-F20; FEI, USA). The carbon and sulfur contents of the prepared samples after the leaching treatment were analyzed by using an infrared carbon-sulfur analyzer (EMIA-920V2; HORIBA, Japan). Additionally, the fractions of CO and CO2 generated during the reduction process were monitored by an infrared gas analyzer (XLZ-1090; XILINZI, China).
Results
Figure 2 depicts the XRD patterns of the products obtained at different temperatures at a MoS2:CaO:C molar ratio of 1:4:1.7, from which it can be seen that, when the mixed samples were annealed at 800°C, 900°C, 1000°C, and 1100°C for 2 h, there are still distinct diffraction peaks of MoS2. In addition, the peaks of CaS, CaMoO4, and Mo were identified. When the reaction temperature rose from 800°C to 1200°C, the diffraction peak intensity of MoS2 gradually reduced and eventually disappeared. Additionally, with the increasing reaction temperature, the diffraction peak of CaMoO4 first increased and then decreased until it disappeared, indicating that CaMoO4 exists as an intermediate phase. Moreover, the diffraction peak intensity of Mo gradually increased. More importantly, it can be seen from Fig. 2 that, when the temperature was increased to 1200°C, the molybdenum-containing phase was only Mo, and the sulfur contained in MoS2 was fixed in CaS. Consequently, a high temperature is favorable for the formation of Mo, and thus the temperature for the preparation of Mo powder was set to 1200°C in the following experiments.

XRD patterns of the products acquired with a MoS2:CaO:C molar ratio of 1:4:1.7 at different temperatures.
In order to investigate the reaction process and determine the optimal reaction time, experiments at 1200°C for 10 min, 20 min, 30 min, and 60 min were conducted, and the corresponding XRD patterns of the samples are shown in Fig. 3a, which shows that the diffraction peaks of MoS2 disappeared even if the sample was only reacted for 10 min, revealing that a high temperature can greatly facilitate the reaction rate. It must be noted that the Mo2C phase was identified when the sample was reacted for 10 min. In addition, CaMoO4 presented when the reaction time was 10 min and 20 min, but disappeared when the reaction time was further prolonged to 30 min and 60 min. To quantitatively analyze the purity of the prepared Mo powder, the residual sulfur and carbon contents of the synthesized Mo powder were evaluated, as shown in Fig. 3b, which it can be seen that both the carbon content and the residual sulfur content decreased with the increase of reaction time. From 10 min to 30 min, the S content decreased from 1.24% to 0.5%, while the C content dropped from 1.53% to 0.25%. However, the C and S contents were almost unchanged when the reaction time was further extended from 30 min to 60 min. Therefore, in order to ensure a high purity of the synthesized Mo powder, the reaction time was set to 60 min.

(a) XRD patterns of the products obtained at 1200°C for different reaction times with a MoS2:CaO:C molar ratio of 1:4:1.7. (b) Carbon and sulfur contents of the samples in (a). (c) XRD patterns of the products acquired at 1200°C for 60 min by adding different amounts of carbon black. (d) Carbon and sulfur contents for the samples obtained at different carbon additions. (e) XRD patterns of the products obtained at 1200°C for 60 min by adding different amounts of CaO. (f) Carbon and sulfur contents of the samples acquired at different CaO additions.
When carbon was used as the reducing agent, the gaseous product contained both CO and CO2. Therefore, the amount of required carbon black should be between the theoretical values corresponding to the generation of CO2 and CO. In addition, to study the effect of the addition amount of carbon black, 2 times the theoretical value of CaO were added, and the molar ratios of MoS2:CaO:C were set to 1:4:1, 1:4:1.5, 1:4:1.6, 1:4:1.7, 1:4:1.8, and 1:4:2, respectively. The XRD patterns of the samples obtained with different amounts of carbon black are shown in Fig. 3c, and indicate that the MoS2 has completely disappeared. With the increase of carbon black content, the diffraction peak intensity of CaMoO4 gradually decreased, and entirely disappeared when the MoS2:CaO:C molar ratio was 1:4:1.6. However, the diffraction peak of Mo2C appeared when the molar ratio of MoS2:CaO:C was 1:4:1.8, and its peak intensity increased when the molar ratio of MoS2:CaO:C was increased to 1:4:2, which indicated that part of the Mo has been carbonized to Mo2C by the excess carbon black. Additionally, almost no CO2 will be formed at 1200°C according to the Boudouard equilibrium (Fig. S2). However, it must be pointed out that the samples were gradually heated to 1200°C, which makes it possible to generate CO2 during the heating process, especially in the low-temperature stage. Therefore, it is reasonable to choose a MoS2:C molar ratio of less than 2 according to the law of mass balance.
The residual sulfur and carbon contents of the products acquired at different MoS2:CaO:C (1:4:1.6, 1:4:1.7, and 1:4:1.8) molar ratios are shown in Fig. 3d. It can be seen that the residual sulfur content is about 0.48%, which decreased slightly with the increase of carbon addition, indicating that the amount of carbon black was not a limiting factor affecting the conversion of MoS2 in the current range. In addition, the lowest carbon content is about 0.18% when the molar ratio of MoS2:CaO:C is equal to 1:4:1.7, while the carbon content sharply increased to 0.91% as the molar ratio of MoS2:CaO:C increased to 1:4:1.8. Consequently, the optimal MoS2:CaO:C molar ratio under the current experimental conditions is 1:4:1.7.
In order to investigate the effect of CaO addition on the desulfurization efficiency, the experiments with MoS2:CaO:C molar ratios of 1:2:1.7, 1:3:1.7, 1:4:1.7, and 1:6:1.7 (1, 1.5, 2, and 4 times, respectively, of the theoretical amount of CaO) at 1200°C for 60 min were conducted. Figure 3e shows the XRD patterns of the different products from which it can be seen that most of the MoS2 was transformed to Mo, with the main co-product of CaS. Additionally, there was still a distinct diffraction peak of MoS2 when the MoS2:CaO:C molar ratio was 1:2:1.7. However, when the MoS2:CaO:C molar ratios were 1:3:1.7, 1:4:1.7, and 1:6:1.7, the diffraction peak of MoS2 disappeared, showing that almost all the MoS2 was fully reacted. Moreover, the C and S contents of the synthesized samples at different CaO additions after leaching treatment were measured, with the results shown in Fig. 3f, from which it can be seen that there was a high residual sulfur content (about 3.98%) when the theoretical amount of CaO was added, and that the residual sulfur content decreased as the addition amount of CaO increased. In addition, there was no obvious change in the residual sulfur content (about 0.46%) when the amount of CaO is larger than 2 times the theoretical value. Consequently, the increase in amount of CaO is beneficial for the conversion of MoS2, and 2 times the theoretical value is necessary for desulfurization.
The morphology of the prepared Mo powder after leaching treatment was further investigated by FE-SEM, as shown in Fig. 4a, b, c, d, e, and f. It can be seen from Fig. 4 that the prepared Mo particles have an ellipsoidal structure with s particle size of 0.5–2 μm, and that there is no obvious change in morphology and particle size of the Mo powder obtained after reacting at 1200°C for 60 min when changing the addition amounts of carbon black. Figure 4g shows the typical TEM image of the prepared Mo powder, and the morphology and particle size agree well with that in Fig 4d. The corresponding selected area electron diffraction (SAED) pattern for the obtained Mo is shown in Fig. 4h, and the perfect diffraction spots reveal that the prepared Mo particle has a single-crystal structure.

FE-SEM images of samples prepared at 1200°C for 60 min at different MoS2:CaO:C molar ratios after the leaching treatment of (a) 1:4:1, (b) 1:4:1.5, (c) 1:4:1.6, (d) 1:4:1.7, (e) 1:4:1.8, and (f) 1:4:2. (g) TEM image and (h) SAED pattern of the sample acquired at 1200°C for 60 min at the MoS2:CaO:C molar ratio of 1:4:1.7.
Discussion
In order to investigate the reaction mechanism, extra experiments were conducted. The samples with a MoS2:CaO molar ratio of 1:4 was annealed in an argon atmosphere at 800°C, 1000°C, and 1200°C for 30 min, respectively. The corresponding XRD patterns of the samples acquired at different temperatures are shown in Fig. 5. It can be seen that, when the samples were reacted at 800°C and 1000°C for 60 min, there were still distinct diffraction peaks of MoS2. Meanwhile, the peaks of CaS, CaMoO4, and Mo were identified. When the temperature rose to 1200°C, the diffraction peaks of MoS2 disappeared, indicating that all the MoS2 had been completely reacted. Additionally, it must be noted that CaMoO4 was generated as the unique molybdenum-containing phase during the reaction. The overall reaction can be represented by Eq. 1.

XRD patterns of the products acquired at 800°C, 1000°C, and 1200°C for 60 min with a MoS2:CaO molar ratio of 1:4.
Padilla et al.20 investigated the reaction mechanism of the samples with a MoS2:CaO molar ratio of 1:2 in an N2 atmosphere at different temperatures, and the detailed reaction mechanism was described by Eqs. 2 and 3. However, in the current study, MoO2 did not appear. The changes of Gibbs free energy of Eqs. 1, 2, and 3 are shown in Fig. S3, from which it can be clearly seen that the changes of Gibbs free energy are all negative in the temperature range of 280–1200°C, indicating that these reactions are all thermodynamically feasible. In Ref. 20, when the CaO/MoS2 molar ratio was 2, there were still strong diffraction peaks of MoS2, MoO2, and CaMoO4 for the samples obtained at 800°C and 1000°C, while no CaO was identified. Therefore, Eqs. 2 and 3 occurred at the same time. When all the S in MoS2 is fixed in the form of CaS, and all the intermediate phase MoO2 is reacted with CaO to generate CaMoO4, the theoretical CaO/MoS2 molar ratio should be 8/3 according to Eq. 1. As can be seen in Fig. 5, when the molar ratio of CaO/MoS2 is equal to 4 (greater than 8/3), strong diffraction peaks of MoS2 were still identified when the samples were prepared at 800°C and 1000°C, indicating that, when the temperature is less than or equal to 1000°C, even if the CaO is excessive, the Eq. 2 cannot proceed completely due to the slow reaction kinetics. However, there is no MoO2 for the sample obtained at 800°C, which demonstrates that all the intermediate phase MoO2 has been reacted according to Eq. 3. Therefore, when the temperature is higher than 800°C and the CaO is sufficient, MoO2 cannot exist in the MoS2-CaO binary system. This proves that the existence of MoO2 in Ref. 20 is caused by the insufficient addition of CaO.
Additionally, another experiment was conducted at 1200°C for 30 min at a MoS2-CaO molar ratio of 1:2, and the XRD results after the reaction are shown in Fig. S4(b). The results are consistent with those in the Ref. 20 (Fig. S4(a)). In Fig. S4(a), the * indicates the unidentified compound, and these diffraction peaks can match well with CaMo6S8 (PDF card. 85-1269), which also shows that the CaO is insufficient to fix the S in the form of CaS under the current conditions. Thus, relative to the current study, the existence of MoO2 in the investigation of Padilla et al. may be due to the insufficient quantity of CaO.20 Additionally, as can be seen from Ref. 20, the MoO2 disappeared when the temperature was raised from 1000°C to 1200°C. Consequently, the temperature and the molar ratio of MoS2:CaO are important factors affecting the generation of MoO2.
Since CaMoO4 was generated as a molybdenum-containing intermediate phase, the reaction mechanism of the CaO-assisted reduction of MoS2 by carbon black can be simplified to the reaction between C and CaMoO4. The binary phase diagrams of CaMoO4-C system under 1 atm and 0.1 atm were calculated by Factsage 7.0, and are shown in Fig. 6. It can be seen that Mo2C and Mo are the possible stable phase when the molar ratio of C:CaMoO4 is in the range of 1–3. In addition, the thermodynamically stable molybdenum-containing phases are Mo2C and CaMoO4 when the temperature is below 1240°C (at 1 atm) or 1080°C (at 0.1 atm). When the temperature is higher than 1240°C or 1080°C, the thermodynamically stable molybdenum-containing phase changes from CaMoO4 + Mo to Mo and then to Mo + Mo2C as the of the molar ratio of C:CaMoO4 increases.

The binary phase diagrams of CaMoO4-C system under different total pressures. (a) P = 1 atm, (b) P = 0.1 atm.
In the actual reaction process, both CO and CO2 were produced. The changes of the volume fractions of the CO and CO2 generated were monitored by an infrared gas analyzer, and the results are shown in Fig. 7. It can be seen that the main gaseous product is CO. From Fig. 3a, it can be seen that the Mo2C phase was identified when the sample reacted at 1200°C for 10 min at a MoS2:CaO:C molar ratio of 1:4:1.7. Additionally, it can be seen from Fig. 3a that Mo2C disappeared as the reaction time increased, which may be due to the reaction between CaMoO4 and Mo2C. This possible reaction can be represented by Eq. 6. Consequently, the main reaction between C and CaMoO4 can be simplify described by Eqs. 4–6.

Changes of volume fractions of CO and CO2 with reaction time.
The changes of Gibbs free energy of Eqs. 4–6 under different partial pressures of CO (PCO) are shown in Fig. S5. It can be seen from Fig. S5(a) that Mo2C can be generated when the temperature is below 1255°C at 1 atm. In this study, argon gas was used as the carrier gas, which greatly decreased the PCO. For instance, if the partial pressure of CO is equal to 0.1 atm, as can be seen in Fig. S5(b), the critical temperature for generating Mo2C is decreased to 1080°C. Therefore, Mo2C is unstable under the current experimental conditions (T = 1200°C, C:CaMoO4 = 2.55), leading to a low carbon content in the prepared Mo powder, as shown in Fig. 3a and b.
According to the experimental results, the main gaseous product is CO. In order to explore the role of CO during the reaction, an extra experiment was performed at 1200°C for 60 min at a MoS2:CaO:C molar ratio of 1:4:1.7. The only difference with the above experiment is that the alumina crucible was covered with an alumina lid to ensure a high partial pressure of CO in the crucible. The XRD result of the sample obtained is shown in Fig. 8. It can be found that the diffraction peak of Mo2C was identified, which illustrates that CO could react with CaMoO4, and the reaction can be represent by reactions 7 and 8. However, in the experiments without alumina lid covered on alumina crucible, it was hard for the generated CO to react with CaMoO4 due to the presence of the carrier gas.

XRD patterns of the product acquired with a lid on the alumina crucible at 1200°C for 60 min at a MoS2:CaO:C molar ratio of 1:4:1.7.
From the above results, it can be found that CaO can be used as an efficient desulfurizer for the molybdenum extraction from molybdenite, and almost all sulfur in MoS2 can be fixed as CaS. Furthermore, a mixture of Mo and CaS can be obtained by adjusting the addition amount of CaO and carbon black, and Mo powder can be acquired after leaching treatment. Additionally, the as-reduced Mo powder is not pure enough for making Mo and its alloys. However, it is possible to use this kind of Mo powder as the molybdenum containing steelmaking additive.
Conclusions
In this work, the molybdenum powder was synthesized via CaO-assisted carbon reduction of MoS2. The following conclusions can be drawn:
-
(1)
During the reaction, MoS2 first reacted with CaO to form CaMoO4, Mo and CaS firstly, and then CaMoO4 was reduced by carbon to generate Mo.
-
(2)
CaO can be used as an efficient desulfurizer for the molybdenum extraction from molybdenite. The addition amount of CaO affects the conversion of MoS2, and the optimal CaO addition is 2 times of the theoretical value.
-
(3)
The main gaseous product is CO, and after reaction almost all sulfur contained in MoS2 can be fixed as CaS.
-
(4)
MoS2 can be almost completely converted to Mo at a MoS2:CaO:C molar ratio of 1:4:1.7 after reacting at 1200°C for 60 minutes. After leaching treatment, the Mo powder with a carbon content of 0.18% and a sulfur content of 0.46% can be acquired.
References
T.A. Lasheen, M.E. El-Ahmady, H.B. Hassib, and A.S. Helal, Min. Proc. Ext. Met. Rev. 36, 145. (2015).
L. Wang, G.H. Zhang, J. Dang, and K.C. Chou, Trans. Nonferr. Metal. Soc. 25, 4167. (2015).
D.H. Wang, G.D. Sun, and G.H. Zhang, Int. J. Refract. Met. Hard Mater. 75, 70. (2018).
A.G. Kholmogorov, O.N. Kononova, G.L. Pashkov, S.V. Kachin, O.N. Panchenko, and O.P. Kalyakina, Euro. J. Miner. Environ. Proc. 2, 82. (2002).
E. Blanco, H.Y. Sohn, G. Han, and K.Y. Hakobyan, Metall. Mater. Trans. B 38, 689. (2007).
Q.S. Zhou, W.T. Yun, J.T. Xi, X.B. Li, T.G. Qi, G.H. Liu, and Z.H. Peng, Trans. Nonferr. Metal Soc. 27, 1618. (2017).
P.V. Aleksandrov, A.S. Medvedev, M.F. Milovanov, V.A. Imideev, S.A. Kotova, and D.O. Moskovskikh, Int. J. Miner. Process. 161, 13. (2017).
Z.F. Cao, H. Zhong, Z.H. Qiu, G.Y. Liu, and W.X. Zhang, Hydrometallurgy 99, 2. https://doi.org/10.1016/j.hydromet.2009.05.001 (2009).
L. Wang, P. Guo, J. Pang, L. Luo, and P. Zhao, Vacuum 116, 77. (2015).
G.H. Zhang, H.Q. Chang, L. Wang, and K.C. Chou, Int. J. Min. Metal. Mater. 25, 405. (2018).
L. Wang, P.M. Guo, P. Zhao, L.B. Kong, and Z.L. Tian, Vacuum 152, 330. (2018).
M. Kumar, T.R. Mankhand, D.S.R. Murthy, R. Mukhpadhyay, and P.M. Prasad, Hydrometallurgy 86, 56. (2007).
F. Habashi, and R. Dugdale, Metall. Trans. 4, 1865. (1973).
P.M. Prasad, T.R. Mankhand, and P. Suryaprakash Rao, Miner. Eng. 6, 857. (1993).
M.M. Afsahi, M. Sohrabi, R.V. Kumar, and H.A. Ebrahim, Thermochim. Acta 473, 61. (2008).
M.M. Afsahi, R.V. Kumar, M. Sohrabi, and Y.J. Park, Chem. Eng. Process 75, 1. (2014).
T.R. Mankhand, and P.M. Prasad, Metall. Trans. B 13, 275. (1982).
P.S. Rao, and P.M. Prasad, Mater. Trans. JIM 34, 1229. (1993).
P.M. Prasad, T.R. Mankhand, P.S.P. Rao, S.N. Singh, and A.J.K. Prasad, Metall. Mater. Trans. B 33, 345. (2002).
R. Padilla, M.C. Ruiz, and H.Y. Sohn, Metall. Mater. Trans. B 28, 265. (1997).
P.K. Tripathy, and R.H. Rakhasia, Min. Proc. Ext. Metall. Rev. 115, 8. (2006).
S. Ghasemi, M.H. Abbasi, A. Saidi, J.Y. Kim, and J.S. Lee, Ind. Eng. Chem. Res. 50(23), 13340. (2011).
S.G. Najafabadi, M.H. Abbasi, and A. Saidi, Thermochim. Acta 503, 46. (2010).
Acknowledgements
This work was financially supported by the State Key Laboratory of Advanced Metallurgy, University of Science and Technology Beijing, China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chang, HQ., Zhang, GH. & Chou, KC. CaO-Assisted Carbothermal Reduction of MoS2 to Synthesize Molybdenum Powder. JOM 73, 2540–2548 (2021). https://doi.org/10.1007/s11837-021-04719-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-021-04719-6