
Abstract
High-throughput density functional theory (HT DFT) is fast becoming a powerful tool for accelerating materials design and discovery by the amassing tens and even hundreds of thousands of DFT calculations in large databases. Complex materials problems can be approached much more efficiently and broadly through the sheer quantity of structures and chemistries available in such databases. Our HT DFT database, the Open Quantum Materials Database (OQMD), contains over 200,000 DFT calculated crystal structures and will be freely available for public use at http://oqmd.org. In this review, we describe the OQMD and its use in five materials problems, spanning a wide range of applications and materials types: (I) Li-air battery combination catalyst/electrodes, (II) Li-ion battery anodes, (III) Li-ion battery cathode coatings reactive with HF, (IV) Mg-alloy long-period stacking ordered (LPSO) strengthening precipitates, and (V) training a machine learning model to predict new stable ternary compounds.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
A foundational concept in materials science and engineering is the processing/structure/properties paradigm: processing determines structure, which in turn defines observed properties. The close ties between structure and properties are historically evident in the development of models to describe materials behavior (e.g., vacancy and dislocation theory). Currently, ab initio predictive models of properties based on structure are commonplace, which has enabled a growing trend in materials design: experimental synthesis in tandem with predictive modeling to facilitate the optimization of materials properties. The promise of such an approach is to dramatically reduce development time for novel materials with innovative design tools and methods. Indeed, accelerating materials design is the primary goal and motivation for the U.S. Materials Genome Initiative.1
A critical innovation toward accelerating materials design has been accurate and efficient first-principles prediction of materials properties with density functional theory (DFT). Employing only quantum mechanical concepts and little experimental input, DFT allows one to predict properties of crystalline solids such as lattice parameters, magnetic moments, formation energies, band structures, etc. Although the fundamental concepts underlying DFT was developed in the 1960s,2,3 it took 20 years or so for the practical application of the theory with efficient DFT codes and algorithms.4–7 Since then, DFT has been one of the great successes in modeling materials behavior.8 With ever-increasing computational power at lower costs and improvements in computational algorithms, the cpu time to perform DFT calculations has been steadily declining to the point that performing large-scale calculations on the order of tens or hundreds of thousands of structures is possible in a reasonable amount of time. So-called high-throughput (HT) DFT calculations enable the generation of large databases of DFT-predicted materials properties,9 which can accelerate materials design through direct searches of materials with desired properties or the development of higher-level models (e.g., data mining).
Several efforts are underway to generate large-scale HT DFT databases, including the Open Quantum Materials Database (OQMD),10 the Materials Project,11 the Computational Materials Repository,12 and AFLOWLIB.13 We have developed the OQMD, which is an extensive HT DFT database consisting of DFT predicted crystallographic parameters and formation energies for over 200,000 experimentally observed International Crystal Structure Database (ICSD)14,15 and theoretical prototype structures, discussed in more detail in the section titled “The OQMD and DFT Accuracy”. An important feature of the OQMD is the open nature of our database, meaning we will provide access to the complete database without limitation for the community to use, strongly in line with the Materials Genome Initiative.1 In the coming months, we will make the OQMD accessible over the web at http://oqmd.org, with a download option available for the database files themselves and the code to use them.
Since the development and application of the earliest HT DFT database,16 HT DFT has proven to be a successful tool for many and varied materials problems.17–25 In this article, we summarize our own HT DFT efforts, beginning with a description of the OQMD, including a broad comparison of DFT formation energies to experimental values.10 We then provide examples from our work of applying HT DFT to several interesting materials problems. First, we employ the OQMD to search for materials with optimum properties for Li-air battery electrodes,26 Li-ion battery anodes,27 and Li-ion battery cathode coatings reactive with HF.28 Then, we use the formation energies of the OQMD to test whether novel Mg-alloy strengthening long-period stacking ordered (LPSO) precipitates are thermodynamically stable structures.29 Last, we describe the use of OQMD formation energies to train a machine learning model with which potential novel ternary compounds are identified.30 We then conclude with thoughts on the future development of HT DFT databases.
The OQMD and DFT Accuracy
The Open Quantum Materials Database is a collection of consistently calculated DFT total energies and relaxed crystal structures. Using the Vienna Ab-initio Simulation Package (VASP),31,32 DFT calculations have been performed for every unique entry in the ICSD without partial site occupancy and less than 35 atoms in the primitive cell, 32,489 structures as of August 2013.10 The OQMD serves two primary functions: as a large set of data for known structures from which optimum materials can be searched (such as in the following sections: “High-Capacity Conversion Anode Screening”, “Li2O Battery Screening”, and “HF Scavenging Li-Ion Battery Cathode Coatings”) and as an accurate description of the chemical potentials and convex hulls of simple and complex systems from which tests of stability can be readily performed (such as in the following sections: “Searching for New Strengthening Precipitates in Lightweight Mg Alloys” and “Data Mining for Novel Ternary Compounds”). The OQMD is primarily limited by what has been experimentally observed and catalogued in the ICSD (i.e., there may exist novel unexplored systems and compounds which are technologically important, see the “Data Mining for Novel Ternary Compounds” section). Toward resolving this issue, we also include in the database DFT calculations of many unary, binary, and ternary prototype structures. These include, for example, every possible combination of A3B L12 and X2YZ heusler chemistries for over 80 elements. The inclusion of these prototypes in the OQMD provides an approximation for unexplored convex hulls and possible undiscovered compounds as they sample unexplored compositions and systems. The total number of structures in the OQMD, including both ICSD structures and prototypes, as of August 2013 is over 200,000 and is growing every day.
For many materials applications, thermodynamic stability is an important quantity. The long-term stability of γ′ Co3(Al,W) L12 precipitate in Co-based superalloys,33 hydrogen storage decomposition pathways in metal borohydrides,34,35 and spinodal decomposition in IV–VI rock salt thermoelectric semiconductors36 are several examples where the stability of phases is critical for understanding materials behavior. For a compound to be stable, it must not only be lower in energy than all other compounds at that stoichiometry but also be lower in energy than linear combinations of all other compounds in a given system. Thus, an accurate description of stability requires the calculation of the phase in question (e.g., Co3 (Al,W)) and all other competing phases in the given system (e.g., Co, CoAl, and Co3W33). Because both the composition and the free energy are linear as a function of quantity of different phases in a system, the set of phases that has the minimum total free energy at a given composition can be determined by linear programming. We have employed this approach, grand canonical linear programming (GCLP),37 to study hydrogen storage reactions,37,38 Li-battery anode conversion materials,27 and general multiphase ground state stability.10 We have recently revised GCLP to make it more efficient when examining stability in highly multicomponent systems.39
As an example of the broad application of GCLP with the DFT-calculated formation energies in the OQMD, we used GCLP to determine how many ICSD compounds in the OQMD are thermodynamically stable at 0 K and zero pressure. Under these conditions, 23% of the 32,489 calculated ICSD structures are stable. Figure 1 shows the quantity of total ICSD structures and OQMD-predicted stable structures by year of their discovery. Before about 1960, most discovered compounds were also thermodynamically stable. After 1960, the number of metastable structures per year grew rapidly, outpacing the fairly constant rate of stable structure discoveries. The surge in thermodynamically metastable yet experimentally observed structures is perhaps due to the advent of complex synthesis techniques for strained and/or high-pressure structures, such as thin-film deposition and the diamond anvil cell.

The number of OQMD-predicted stable compounds and total ICSD compounds by year of their discovery. The year for a structure corresponds to the earliest publication year for ICSD entries at that given structure’s composition and symmetry
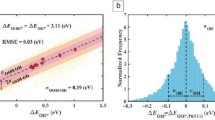
As a large database of DFT calculations, the OQMD can be used to perform a broad comparison of DFT predicted properties to experimental measurements to assess the accuracy of the DFT approach. We have done so for thermodynamic stability by comparing 1290 DFT and experimentally measured formation energies compiled in the Scientific Group Thermodata Europe SSUB thermodynamic database.40 The SSUB does not contain structural information about a given compound, only a composition. Therefore, we compare an SSUB formation energy to the most stable structure at that composition predicted by DFT, as shown in Fig. 2. The average error between DFT and experimental formation energies is 24 meV/atom and the mean absolute error is 113 meV/atom.

Comparison of the DFT-predicted formation energies with difference between the experimental40 and DFT values. The black line indicates perfect agreement between the two, the solid red line indicates the average agreement, and the dashed red lines one and two standard deviations. Histograms are provided for both axes. The thick red line corresponds to a normal distribution fitted to the histogram of the formation energy differences
Results—Successful Applications of OQMD Database
In the following sections, we will discuss five examples from our work for the application of the OQMD to materials design problems covering a range of materials types.
High-Capacity Conversion Anode Screening
Because of their commercial significance and amenability to simple bulk analysis with DFT, batteries were one of the first areas to be tackled using HT DFT.41,42 While previous high-throughput studies have focused on the cathode of Li-ion batteries, we have used the OQMD to search for novel high-capacity anodes27 in three promising classes of materials: transition metal silicides, stannides, and phosphides. Although a patchwork of silicon,43–46 tin,47–50 and phosphorus51–55 compounds has been explored as anode materials in the past, a complete and systematic study of these materials had never been undertaken. We studied this entire class of materials, looking for promising battery reactions based on a series of thermodynamic screens.
Important descriptors for a battery reaction are voltage, gravimetric and volumetric capacity, and volume expansion. The voltage is important in an anode for two reasons: (I) In a low-voltage anode, where the voltage is only slightly higher than that of metallic lithium, lithium metal can form as dendrites, introducing major safety risks, and (II) in a high-voltage anode, the energy density of the total cell is diminished. Finally, it has been observed that internal stresses from the enormous volume expansion associated with extremely high lithium capacity in silicon lead to rapid cell degradation.56 We speculated that finding reactions which have lower volume product phases may represent an avenue to lower volume expansion—and improved cyclability—in anode materials.
All three anode traits described can be calculated from DFT-predicted ground-state thermodynamics. We explored all possible Li-ion anode reactions within the OQMD to find the optimum anode material based on these quantities, as summarized in Fig. 3. As a result of this screening process, several conversion reactions were found that exhibited high capacities, moderate voltages, and minimal volume expansion. As a promising indicator of the selective ability of our screens, one of the anode materials that passed through the screen was CoSn—which is already a known anode material,57 currently employed as the active component of Sony’s Nexelion battery (Sony Corporation, Tokyo, Japan). Sixteen potential candidate reactions were predicted, three of which stood out as being the most promising: TiP, LiSiNi2, and CoSi2.

Initial voltage and gravimetric capacity for transition metal silicides, stannides, and phosphides lithiation reactions in the OQMD. The color of the points is determined by the volume change per lithium atom for the reaction
Li2O Battery Screening
Recently, a new approach to lithium-air batteries was described by Johnson et al.58 and Trahey et al.59 in which Li5FeO4 was cycled in a cell open to oxygen. In this system, after electrochemically delithiating this material, a surprisingly high voltage was observed. This high voltage covers a large amount of capacity and is attributed to the reinsertion of Li2O units. When such novel reaction types or battery architectures are developed, it is tremendously efficient to survey the new field very rapidly using a HT DFT database because the new search space is so open to exploration. To that end, the existing database of calculated structures in the OQMD was employed to screen for other reactions involving Li2O units that may follow a similar path.26
To begin, the OQMD was used to define every possible reaction of structures which satisfies the equation
where for the completeness of the search, “A” can be any compound and not just an element. For instance, Li4KAlO4 in this reaction can react to form KAlO2. According to the search, 255 “A” compounds in the ICSD can satisfy Eq. 1, as summarized in Fig. 4. The stability of the compounds is then taken into account, excluding reactions that do not occur by two-phase equilibria (i.e., indirect).37,60 Last, several of additional constraints were applied to mimic the constraints on real battery materials. Reactions were terminated (I) when the reaction encountered a material with a wide DFT bandgap; (II) when the next decomposition step occurs at a high voltage, such that it might endanger the electrolyte; or (III) if the reaction passes through a region of phase equilibria containing more than two phases.

Initial voltage and gravimetric capacity for HT DFT predicted Li-ion anode reactions (Eq. 1). The color of each point indicates the widest DFT bandgap of any step in the reaction, with red indicating wide bandgap reactions and blue indicating narrow bandgap reactions. The shape of the points indicates either direct (circles) or indirect (crosses) reaction paths
This reaction enumeration process was performed under a set of very stringent requirements and again at a wider tolerance. Once the reactions were determined, the results were screened for the highest possible capacity. The best materials that were identified by the very stringent requirements are being disclosed in another publication,26 but among the rest of the results were several known expected reactions (which builds our confidence in the screening methodology), as well as several new, promising, and previously unknown reactions. For instance, the reaction that was the inspiration for this search (Li5FeO4) was recovered, which supports the selection of reaction descriptors and screening criteria. Several more reactions show potential when the less strict screening conditions are used. Among these is LiOH, which is the reaction product of Li-water cells,35,61,62 which have been observed. Also predicted is Li2CO3; it is the dead product phase of CO2 and lithia, which is the reason CO2 must be scrubbed from Li-air cells, and has also been studied as a possible battery itself.63 As a result of this study, the list of reactions that are likely worth experimental investigation has been reduced from 255 to the 10 most likely to yield high capacity and cyclability.26
HF Scavenging Li-Ion Battery Cathode Coatings
Degradation of electrodes, induced by their contact with the electrolyte, has to be suppressed to improve the capacity retention and rate capability of Li-ion batteries.64,65 Coating the cathode material is an effective remedy to retard the capacity fading upon cycling,66,67 but experimentally testing all coating candidates for a given cathode is a resource-demanding task that requires fabrication, cycling, and disassembly of the batteries. We have recently performed an HT DFT screening of metal oxide cathode coatings for Li-ion batteries to predict promising coatings a priori.28 The hydrogen-fluoride (HF) scavenging capability of the coating materials was chosen as the primary design attribute because the highly corrosive HF present in conventional LiPF6-based electrolytes is known to attack the cathode and trigger the dissolution of redox-active metal ions into the electrolyte.64,68 Contemporary battery technology requires functional coatings that can preferentially react with HF in presence of the cathode,69 beyond what is expected from a simple protective barrier. We employ the OQMD to scan for materials that can serve as HF scavengers.28
For an oxide coating \( \left( {{\text{M}}_{x} {\text{O}}_{\frac{1}{2}} } \right), \) a generic HF-scavenging reaction producing its conjugate fluoride (M x F) can be written as
The free energy of this reaction, approximated as the DFT enthalpy at T = 0 K (∆H s-HF), is a natural measure of the HF-scavenging capability of the coating \( {\text{M}}_{x} {\text{O}}_{\frac{1}{2}} \). The first screening criterion was selected as ∆H s-HF [coating] < ∆H s-HF [cathode] to ensure the preferential reaction of HF with the coating over the cathode material. However, to estimate AH s-HF [cathode], a reaction between the cathode material and HF (i.e., the HF-attack reaction) analogous to Eq. 2 must be devised, such as \( \frac{1}{4}{\text{LiCoO}}_{ 2} + {\text{ HF}} \to {\text{Product}}\left( {\text{s}} \right) + \frac{1}{2}{\text{H}}_{ 2} {\text{O}} \) for LiCoO2, where one needs to specify the product(s). Our approach to this problem was to find the combination of phases that yields the lowest energy at the chemical composition corresponding to the product(s) in the HF-attack reactions using GCLP along with OQMD. Because the calculated ∆H s-HF [cathode] values is on the order of −0.3 eV/HF for typical cathode materials (LiCoO2, LiNiO2, LiMn2O4, LiFePO4, etc.), the first screen amounts to finding coating materials with ∆H s-HF values more negative than –0.3 eV/HF. Volumetric (ΩV) and gravimetric (ΩG) HF-scavenging capacities (defined as moles of HF that a coating scavenges per unit volume and gram, respectively) were introduced as additional design parameters. As the volume of the coating increases, it becomes more likely to impede Li and electron transport, and as the mass of the coating increases, specific properties (such as the energy density) degrade. Consequently, to design optimal coatings, both ΩV and ΩG need to be maximized.
Unlike the insertion cathodes with fast kinetics, stable oxides and fluorides are expected to be lithiated mostly via relatively sluggish conversion reactions70,71 requiring significant overpotentials to reverse upon charging. The overpotential may lead to entrapment of Li in the coating and impair the rate capability as well as the capacity of the battery. This cyclable-Li loss into a coating may occur if the voltage decreases to a level comparable to the lithiation voltage of the coating upon discharging the battery. Accordingly, we selected a typical Li-ion battery discharge cutoff of ~3.0 V as the upper lithiation voltage limit for screening the coatings. In fact, the conjugate fluoride M x F of a given oxide \( {\text{M}}_{x} {\text{O}}_{\frac{1}{2}} \) in Eq. 2 almost always has a conversion voltage higher than \( {\text{M}}_{x} {\text{O}}_{\frac{1}{2}} \) (except for a few alkali/alkaline earth M).28 Fluorides are, therefore, more prone to reacting with lithium, and accordingly, the lithiation voltage of \( {\text{M}}_{x} {\text{F}} \) was chosen as the fourth design attribute.
The high-throughput screening was carried out within the four-dimensional design space of ΔH s-HF, ΩV, ΩG, and lithiation voltage in a set of 81 s-, p-, and d-block binary metal oxide coating candidates, for which a reaction in the form of Eq. 2 can be devised using the compounds available in the ICSD (see Fig. 5). Materials that are experimentally known to be effective coatings, such as Al2O3, ZrO2, and MgO,67–74 passed through all screens, and Al2O3 was found to provide an optimal compromise among all four attributes. Besides this validation of the selected design parameters, we observed that the extent of the experimental capacity retention provided by different coatings on the same cathode is correlated with their HF-scavenging tendencies and capacities, as long as the fluoride produced by the HF-scavenging reaction is a stable solid near room temperature. With an HT thermodynamic analysis of 81 binary oxides, we predicted several new, promising cathode coating candidates with attributes similar to the well-tested coating materials such as Al2O3 and MgO (see Fig. 5).

Calculated HF-scavenging tendency (–ΔH s-HF) of binary metal oxides versus lithiation voltage of the corresponding metal fluoride product layers. Dashed lines enclose the compounds that pass the –ΔH s-HF and lithiation voltage screens. Point sizes are proportional to the gravimetric HF-scavenging capacities
Searching for New Strengthening Precipitates in Lightweight Mg Alloys
A critical approach to improve the efficiency of transportation systems is to reduce the weight of the vehicles, where a 10% reduction in the weight of a conventional combustion automobile can improve fuel efficiency by 6–8%.1 As one of the lightest structural metals, Mg and Mg-based alloys offer an attractive alternative to Al and steel vehicle parts. However, issues with poor strength and ductility have limited the use of Mg in automobiles. Therefore, there has been a large effort in recent years to improve the mechanical properties of Mg alloys. Rare earth (RE) solute additions improve Mg alloy strength and ductility through the appearance of novel precipitates and strengthening mechanisms.75 One such precipitate, long-period stacking ordered (LPSO) structures, is responsible for dramatic increases in yield strength and ductility, 610 MPa at 15% elongation.76 However, requiring as much as 1 at.% of expensive RE elements, Mg alloys containing LPSO structures are too costly for many industrial applications. Therefore, we employed HT DFT and the OQMD database to search for alternative, more affordable LPSO-forming elements.29
LPSO structures are observed to be Mg-rich ternary precipitates in Mg-XL-XS ternary systems,77–87 where XL is an element larger than Mg and XS is an element smaller than Mg. LPSO structures have been observed in several ternary systems, notably Mg-Y-Zn, as summarized in Fig. 6. LPSO structures, as the name implies, are precipitates that exhibit long-period order of atomic layer stacking along the Mg hexagonal close-packed (hcp) c-axis. The structures contain order of both the stacking of atomic layers, which alternate between hcp- and face-centered cubic (fcc)-type stacking, as well as chemical order within the fcc-stacked layers, with fully ordered arrangements of binary and ternary sets of elements.88,89 18R and 14H LPSO structures have been observed,81,90 where the number corresponds to the number of atomic layers in the period of the c-axis stacking, and the letter refers to whether the structure has rhombohedral or hexagonal symmetry. Only recently has the crystal structure of the LPSO precipitates been fully determined,88,89 with Mg71X L8 X S6 as the formula for the 14H LPSO “interstitial” structure model.91 For a DFT study of intermetallic stability, where energy differences are on the order of 0.01 eV/atom, an accurate crystal structure is necessary.

DFT predicted stability of 14H-i and 18R-i LPSO structures for Mg-XL-XS ternary systems. XS and XL elements are given along the vertical and horizontal axes, respectively. The colors are defined by the stability of the LPSO structure relative to the convex hull: blue if the LPSO structure is on the convex hull, yellow if it is within 25 meV/atom above the convex hull, and red if it is more than 25 meV/atom above the convex hull. XL = RE systems are given at top and XL ≠ RE systems at bottom. Experimentally observed LPSO-forming systems are also indicated with triangles.77–87 Blue (and possibly yellow) squares without triangles represent predictions of alloy systems where as yet unobserved LPSO structures should be stable
To predict novel non-RE LPSO chemistries, we first tested the ability of DFT to predict the known LPSO-forming Mg-XL-XS ternary systems. The stabilities of 85 Mg-RE-XS LPSO structures were predicted with DFT by comparing the LPSO structure total energy to the OQMD convex hulls for every system as generated by GCLP at the LPSO composition. The results, summarized in Fig. 6, agree perfectly with experimental observations, where all 11 known LPSO forming ternary systems are predicted by DFT to form a stable LPSO structure. Furthermore, 41 novel RE-containing LPSO systems are predicted as well, each representing a ternary system awaiting experimental investigation and confirmation of the DFT prediction. Having proven DFT’s ability to predict LPSO stability, we extended the search to include 11 non-RE XL elements, primarily focusing on elements larger than Mg. As shown in Fig. 6, four non-RE elements are shown to form stable, or nearly stable, LPSO structures: Pa, Ca, Th, and Sr. Pa and Th are radioactive, severely limiting their applicability. Ca and Sr are promising, particularly the Mg-Ca-Zn system, which is predicted to form stable LPSO structures. Mg-Ca-Zn alloys have been explored experimentally and LPSO structures have not been observed.92–95 However, these alloys have different compositions from those that have formed LPSO structures and were not extruded in the manner that has known to readily form LPSO structures. Therefore, using HT DFT and the OQMD, we predict not only 41 novel RE-containing LPSO systems but also Ca and Sr additions as potential affordable alternatives to RE elements to form LPSO structures in Mg alloys.
Data Mining for Novel Ternary Compounds
An issue with HT DFT materials design is that the set of materials in the database is limited to those experimentally observed because DFT requires, as input, the crystal structure. There are two consequences of this limitation. First, the true convex hull for unexplored systems may be incorrect. To an extent, this is addressed within the OQMD in a limited way by the calculation of many prototype structures, as discussed in the section titled “The OQMD and DFT Accuracy”. Second, the discovery of novel materials is limited to the set of structures calculated within the database. For example, a search for novel large bandgap materials would be limited to structures observed in the ICSD and common prototypes as opposed to truly novel complex crystal structures. Predicting the ground-state crystal structure for arbitrary compositions remains a heavily studied yet elusive goal.38,96–98 The primary difficulty is that, at a given composition, many structures (on the order of millions) must be tested to ensure an accurate prediction of the ground state. For sufficient accuracy, DFT must be used to test the quality of potential structures but is still far too costly for this task.
Meredig et al.30 approached this problem by avoiding the question of structure altogether by training a pair of heuristic and machine learning (ML) models with OQMD data and using a combination of the models to quantitatively predict the formation energies of arbitrary ternary compositions. With such an approach, all possible ternary compositions can be explored at a fraction of the computational time and cost of a single DFT calculation and the most likely compositions with novel ternary compounds can be explored with DFT structure prediction methods or experiments. The heuristic model employs a well-established approach to predict A-B-C ternary formation energies by taking the composition-weighted average of those from the A-B, A-C, and B-C binary systems.99,100 When applied to the known OQMD data, this heuristic greatly underestimates the DFT formation energies but in a very systematic way. Fitting the heuristic to the DFT calculated OQMD ternary formation energies results in a simple correction: AH heur-corrF = 1.50ΔH heurF − 0.02 eV. The ML model101 incorporates numerous decision trees to learn the behavior of element interactions to quantitatively predict formation energies of arbitrary compositions. Although ML has been previously used to predict stable crystal structures at given compositions,97,102,103 the application of ML in the current work is unique in that a quantitative, structure-independent property prediction is produced.
The accuracy of the combined heuristic and machine learning model approach has been tested by training the models with 4000 OQMD ternary compound formation energies and then using the models to predict the formation energies of ~8700 others that have been already calculated within OQMD. The agreement with DFT is very close, with mean absolute errors about half of what is typical of DFT compared to experiment (~0.11 eV/atom, see the section titled “The OQMD and DFT Accuracy”). Having demonstrated the ability of these two models to predict the formation energy of ternary compounds without the need for an input structure, Meredig et al.30 then applied these models to predict the formation energies of over 1.6 million ternary compositions. These results are summarized in Fig. 7, where the likelihood of a given pair of elements producing a stable ternary compound is plotted, as determined by ranking the stabilities of all the predicted compositions. Such a ranking is made possible because the combined heuristic-ML approach can predict the stabilities of the 1.6 million compositions in minutes, whereas a DFT ground state search of each composition would require tens of thousands of cpu-years. Last, the nine most likely ternary compositions to form new compounds were investigated further with DFT crystal structure prediction to determine possible stable ground states. Of the nine compositions, eight of them form new stable compounds: SiYb3F5, Pa2O(SiO6), U2O(PO4)2, S2(VF6), Pm2S3, P3(BrCs4), Te3Y4N2, and Ba(TeS3). There are, of course, many more potential compositions to be explored (approximately 4500), and this approach, currently demonstrated with formation energy, can be applied to many other materials properties for efficient and broad materials discovery. Thus, machine learning and heuristic models trained on OQMD data have been used to efficiently predict several thousand novel stable ternary compositions.

Heat map of 1.6 M candidate ternary compositions’ stability rankings according to the combined heuristic-ML model. Brighter colors imply higher rankings (greater stability). Each point on the heat map corresponds to the average likelihood of stability of all ternary compounds containing the two elements on the plot axes; e.g., the Fe-Cl point gives the average likelihood of stability of all Fe-Cl-X compounds. The black bars on the plot correspond to either noble gases or several exotic heavy elements that were not considered in the survey
Future Outlook
High-throughput density functional theory is fast becoming a powerful tool for approaching complex materials design problems. We have summarized several examples where we successfully employed the OQMD in materials discovery, data mining, and materials optimization. Progress for the creation and use of HT DFT databases will continue along two paths: increasing complexity and increasing understanding. As to the former, the ever-improving speed and efficiency of computing processors will allow for more accurate and more costly calculations to be performed. Both with predictive models more accurate than DFT (such as the GW approximation,104 the random phase approximation,105 and quantum Monte Carlo106) and more elaborate DFT calculations (such as finite-temperature frozen phonon calculations and larger, more complex prototype structures), the prediction of HT DFT will grow more accurate and be applicable to more materials properties over time. We are advancing OQMD in this regard with the calculation of more complex and higher-order prototype compounds, including perovskite and heusler structures for every possible chemistry, elastic tensor calculations of the ICSD structures, and the thermodynamic and electronic effects of dilute mixing in thermoelectric materials, all of which will be openly available in updates to the OQMD.
The second path of the future of HT DFT, increasing understanding, is likely the more challenging of the two. With the increasing capability to create new data, making sense of it all becomes more difficult. Data mining techniques, such as those demonstrated in the “Data Mining for Novel Ternary Compounds” section, will become critical to effectively solve materials problems with HT DFT databases such as OQMD. Our own future efforts in this regard include complex chemical potential fits to more accurately describe the formation energy of transition metal oxides and halides (compared to the simple fits employed in “The OQMD and DFT Accuracy” section). We are also developing additional machine learning models for new types of materials properties, such as bandgap, magnetic moment, and vacancy formation energy. The breadth of HT DFT calculations is also a challenge because tools such as GCLP are only efficient for investigating phase stability in specific, small regions of composition space. We are building tools to efficiently explore and analyze ground state thermodynamics for broad searches of composition space (e.g., 10-component phase diagrams). With improved DFT calculations and data analysis tools, HT DFT will become an even more critical tool in materials science.
References
National Science and Technology Council, Materials Genome Initiative for Global Competitiveness, Tech. Rep. (June 2011).
P. Hohenberg and W. Kohn, Phys. Rev. 136, B864 (1964).
W. Kohn and L.J. Sham, Phys. Rev. 140, A1133 (1965).
J. Perdew and A. Zunger, Phys. Rev. B 23, 5048 (1981).
J. Ihm, A. Zunger, and M.L. Cohen, J. Phys. C Solid State 12, 4409 (1979).
D. Ceperley and B. Alder, Phys. Rev. Lett. 45, 566 (1980).
J. Ihm, M. Yin, and M.L. Cohen, Solid State Commun. 37, 491 (1981).
J. Hafner, C. Wolverton, and G. Ceder, MRS Bull. 31, 659 (2006).
S. Curtarolo, G.L.W. Hart, M.B. Nardelli, N. Mingo, S. Sanvito, and O. Levy, Nat. Mater. 12, 191 (2013).
J. Saal, S. Kirklin, B. Meredig, A. Thompson, J. Doak, and C. Wolverton, unpublished research (2013).
A. Jain, G. Hautier, C.J. Moore, S. Ping Ong, C.C. Fischer, T. Mueller, K.A. Persson, and G. Ceder, Comput. Mater. Sci. 50, 2295 (2011).
D.D. Landis, J.S. Hummelshoj, S. Nestorov, J. Greeley, M. Dulak, T. Bli-gaard, J.K. Norskov, and K.W. Jacobsen, Comput. Sci. Eng. 14, 51 (2012).
S. Curtarolo, W. Setyawan, S. Wang, J. Xue, K. Yang, R.H. Taylor, L.J. Nelson, G.L. Hart, S. Sanvito, M. Buongiorno-Nardelli, N. Mingo, and O. Levy, Comput. Mater. Sci. 58, 227 (2012).
G. Bergerhoff, R. Hundt, R. Sievers, and I.D. Brown, J. Chem. Inf. Model. 23, 66 (1983).
A. Belsky, M. Hellenbrandt, V.L. Karen, and P. Luksch, Acta Crystallogr. B 58, 364 (2002).
G. Jóhannesson, T. Bligaard, A. Ruban, H. Skriver, K. Jacobsen, and J. Norskov, Phys. Rev. Lett. 88, 1 (2002).
T. Bligaard, G.H. Johannesson, A.V. Ruban, H.L. Skriver, K.W. Jacobsen, and J.K. Nørskov, Appl. Phys. Lett. 83, 4527 (2003).
M. Andersson, T. Bligaard, A. Kustov, K. Larsen, J. Greeley, T. Johannessen, C. Christensen, and J. Nørskov, J. Catal. 239, 501 (2006).
J. Greeley, T.F. Jaramillo, J. Bonde, I.B. Chorkendorff, and J.K. Norskov, Nat. Mater. 5, 909 (2006).
A. Jain, S.-A. Seyed-Reihani, C.C. Fischer, D.J. Couling, G. Ceder, and W.H. Green, Chem. Eng. Sci. 65, 3025 (2010).
G. Hautier, A. Jain, S.P. Ong, B. Kang, C. Moore, R. Doe, and G. Ceder, Chem. Mater. 23, 3495 (2011).
S. Wang, Z. Wang, W. Setyawan, N. Mingo, and S. Curtarolo, Phys. Rev. X 1, 021012 (2011).
K. Yang, W. Setyawan, S. Wang, M. Buongiorno Nardelli, and S. Curtarolo, Nat. Mater. 11, 614 (2012).
L. Yu and A. Zunger, Phys. Rev. Lett. 108, 068701 (2012).
X. Zhang, L. Yu, A. Zakutayev, and A. Zunger, Adv. Funct. Mater. 22, 1425 (2012).
S. Kirklin and C. Wolverton, unpublished research (2013).
S. Kirklin, B. Meredig, and C. Wolverton, Adv. Energy Mater. 3, 252 (2013).
M. Aykol, S. Kirklin, and C. Wolverton, unpublished research (2013).
J. Saal and C. Wolverton, unpublished research (2013).
B. Meredig, A. Agrawal, S. Kirklin, J.E. Saal, J.W. Doak, A. Thompson, K. Zhang, A. Choudhary, and C. Wolverton, unpublished research (2013).
G. Kresse and J. Furthmuller, Phys. Rev. B 54, 11169 (1996).
G. Kresse and J. Furthmuller, Comput. Mater. Sci. 6, 15 (1996).
J. Saal and C. Wolverton, Acta Mater. 61, 2330 (2013).
Y. Zhang, E. Majzoub, V. Ozoliņš, and C. Wolverton, J. Phys. Chem. C 116, 10522 (2012).
Y. Zhang, E. Majzoub, V. Ozoliņš, and C. Wolverton, Phys. Rev. B 82, 174107 (2010).
J.W. Doak and C. Wolverton, Phys. Rev. B 86, 144202 (2012).
A. Akbarzadeh, V. Ozolins, and C. Wolverton, Adv. Mater. 19, 3233 (2007).
C. Wolverton and V. Ozolins, Phys. Rev. B 75, 1 (2007).
S. Kirklin and C. Wolverton, unpublished research.
SGTE, Thermodynamic Properties of Inorganic Materials, vol. 19 (Berlin, Germany: Springer-Verlag, 1999).
Y. Meng and M.A.D. Dompablo, Energy Environ. Sci. 2, 589 (2009).
G. Ceder, MRS Bull. 35, 693 (2010).
M.D. Fleischauer, J.M. Topple, and J.R. Dahn, Electrochem. Solid State 8, A137 (2005).
H. Lee, Y. Kim, M. Hong, and S. Lee, J. Power Sources 141, 159 (2005).
C.-M. Hwang and J.-W. Park, Surf. Coat. Technol. 205, S439 (2010).
Z. Wang, W. Tian, X. Liu, Y. Li, and X. Li, Mater. Chem. Phys. 100, 92 (2006).
A.D.W. Todd, R.E. Mar, and J.R. Dahn, J. Electrochem. Soc. 154, A597 (2007).
M. Xue and Z. Fu, Solid State Ionics 177, 1501 (2006).
P. Ferguson and M. Martine, J. Power Sources 194, 794 (2009).
X. Wang and W. Han, ACS Appl. Mater. Int. 2, 1548 (2010).
J. Xiang, X. Wang, X. Xia, J. Zhong, and J. Tu, J. Alloy. Compd. 509, 157 (2011).
D. Souza, V. Pralong, A. Jacobson, and L. Nazar, Science 296, 2012 (2002).
V. Pralong, Electrochem. Commun. 4, 516 (2002).
J.L. Tirado, J.C. Jumas, L. Monconduit, and J. Olivier-Fourcade, J. Power Sources 109, 308 (2002).
S. Boyanov, F. Gillot, and L. Monconduit, Ionics 14, 125 (2008).
L.Y. Beaulieu, K.W. Eberman, R.L. Turner, L.J. Krause, and J.R. Dahn, Electrochem. Solid State 4, A137 (2001).
J. Wolfenstine, J.L. Allen, J. Read, and D. Foster, Tech. Rep. June (2006).
C.S. Johnson, S.-H. Kang, J.T. Vaughey, S.V. Pol, M. Balasubramanian, and M.M. Thackeray, Chem. Mater. 22, 1263 (2010).
L. Trahey, C. Johnson, and J. Vaughey, Electrochem. Solid State 14, A64 (2011).
C. Wolverton, D.J. Siegel, A.R. Akbarzadeh, V. Ozoliņš, J. Phys.-Condens. Mater. 20, 064228 (2008).
E. Littauer, W. Momyer, and K. Tsai, J. Power Sources 2, 163 (1977).
M. Urquidi-Macdonald, J. Flores, D. Macdonald, O. Pensado-Rodriguez, and D. Vanvoorhis, Electrochim. Acta 43, 3069 (1998).
K. Takechi, T. Shiga, and T. Asaoka, Chem. Commun. 47, 3463 (2011).
Z. Chen, Y. Qin, K. Amine, and Y.-K. Sun, J. Mater. Chem. 20, 7606 (2010).
K. Edstrom, T. Gustafsson, and J. Thomas, Electrochim. Acta 50, 397 (2004).
Y.J. Kim, J. Cho, T.-J. Kim, and B. Park, J. Electrochem. Soc. 150, A1723 (2003).
J. Cho, Y.J. Kim, and B. Park, Chem. Mater. 4, 3788 (2000).
D. Aurbach, B. Markovsky, A. Rodkin, and E. Levi, Electrochim. Acta 47, 4291 (2002).
N.V. Landschoot, E. Kelder, P. Kooyman, C. Kwakernaak, and J. Schoonman, J. Power Sources 138, 262 (2004).
J. Cabana, L. Monconduit, D. Larcher, and M.R. Palacín, Adv. Mater. 22, E170 (2010).
F. Wang, J. Am. Chem. Soc. 133, 18828 (2011).
J. Cho, T.-G. Kim, C. Kim, J.-G. Lee, Y.-W. Kim, and B. Park, J. Power Sources 146, 58 (2005).
H. Zhao, L. Gao, W. Qiu, and X. Zhang, J. Power Sources 132, 195 (2004).
C. Li, H. Zhang, L. Fu, H. Liu, Y. Wu, E. Rahm, R. Holze, and H. Wu, Electrochim. Acta 51, 3872 (2006).
J.-F. Nie, Metall. Mater. Trans. A 43, 3891 (2012).
Y. Kawamura, K. Hayashi, A. Inoue, and T. Masumoto, Metall. Mater. Trans. 42, 1172 (2001).
K. Amiya, T. Ohsuna, and A. Inoue, Mater. Trans. 44, 2151 (2003).
M. Yamasaki, T. Anan, S. Yoshimoto, and Y. Kawamura, Scripta Mater. 53, 799 (2005).
Y. Kawamura, T. Kasahara, S. Izumi, and M. Yamasaki, Scripta Mater. 55, 453 (2006).
K. Yamada, Y. Okubo, M. Shiono, and H. Watanabe, Mater. Trans. 47, 1066 (2006).
Y. Kawamur and M. Yamasaki, Metall. Mater. Trans. 48, 2986 (2007).
T. Itoi, K. Takahashi, H. Moriyama, and M. Hirohashi, Scripta Mater. 59, 1155 (2008).
J. Nie, K. Ohishi, X. Gao, and K. Hono, Acta Mater. 56, 6061 (2008).
H. Yokobayashi, K. Kishida, H. Inui, M. Yamasaki, and Y. Kawamura, Acta Mater. 59, 7287 (2011).
S.-B. Mi and Q–.Q. Jin, Scripta Mater. 68, 635 (2013).
Q.-Q. Jin, C.-F. Fang, and S.-B. Mi, J. Alloy Compd. 7 (2013).
Z. Leng, J. Zhang, T. Yin, L. Zhang, S. Liu, M. Zhang, and R. Wu, Mater. Sci. Eng.: A 580, 196 (2013).
Y. Zhu, A. Morton, and J. Nie, Acta Mater. 58, 2936 (2010).
D. Egusa and E. Abe, Acta Mater. 60, 166 (2012).
T. Itoi, T. Seimiya, Y. Kawamura, and M. Hirohashi, Scripta Mater. 51, 107 (2004).
D. Egusa and E. Abe (Paper presented at LPSO Conference, Sapporo, Japan, 2 October 2012).
H. Somekawa and T. Mukai, Mater. Sci. Eng.: A 459, 366 (2007).
Y.-N. Zhang, D. Kevorkov, J. Li, E. Essadiqi, and M. Medraj, Intermetallics 18, 2404 (2010).
K. Oh-ishi, R. Watanabe, C. Mendis, and K. Hono, Mater. Sci. Eng.: A 526, 177 (2009).
T. Zhou, M. Yang, Z. Zhou, J. Hu, and Z. Chen, J. Alloy. Compd. 560, 161 (2013).
G. Trimarchi and A. Zunger, Phys. Rev. B 75, 104113 (2007).
C.C. Fischer, K.J. Tibbetts, D. Morgan, and G. Ceder, Nat. Mater. 5, 641 (2006).
C.W. Glass, A.R. Oganov, and N. Hansen, Comput. Phys. Commun. 175, 713 (2006).
Y.M. Muggianu, M. Gambino, and J.P. Bros, J. Chim. Phys. 72, 83 (1975).
M. Hillert, Phase Equilibria, Phase Diagrams and Phase Transformations (Cambridge, U.K.: Cambridge University Press, 1998).
J.J. Rodríguez, L.I. Kuncheva, and C.J. Alonso, IEEE Trans. Pattern Anal. Mach. Intell. 28, 1619 (2006).
G. Hautier, C.C. Fischer, A. Jain, T. Mueller, and G. Ceder, Chem. Mater. 22, 3762 (2010).
S. Curtarolo, D. Morgan, K. Persson, J. Rodgers, and G. Ceder, Phys. Rev. Lett. 91, 135503 (2003).
M. Shishkin and G. Kresse, Phys. Rev. B 75, 235102 (2007).
J. Harl and G. Kresse, Phys. Rev. Lett. 103, 056401 (2009).
W. Foulkes, L. Mitas, R. Needs, and G. Rajagopal, Rev. Mod. Phys. 73, 33 (2001).
Acknowledgements
We would like to acknowledge funding support from the following sources: the Center for Electrical Energy Storage: Tailored Interfaces, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (to S. K.); The Dow Chemical Company (to M. A.); the Ford-Boeing-Northwestern Alliance for the LPSO study and U.S. Department of Energy, Office of Basic Energy Sciences through grant DE-FG02-98ER45721 for work on the OQMD (to J. E. S.); and the Department of Defense through the National Defense Science & Engineering Graduate Fellowship Program with further support by DOE under Grant No. DE-FG02-07ER46433 (to B. M. and C. W.). Calculations were performed on the Northwestern University high-performance computing system Quest, as well as on resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Saal, J.E., Kirklin, S., Aykol, M. et al. Materials Design and Discovery with High-Throughput Density Functional Theory: The Open Quantum Materials Database (OQMD). JOM 65, 1501–1509 (2013). https://doi.org/10.1007/s11837-013-0755-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-013-0755-4