
Abstract
Tobacco bacterial wilt caused by Ralstonia solanacearum invades tobacco plants during the whole growth period affecting yield and quality. However, the transcriptome profiling of tobacco plant in response to bacterial wilt has not been well studied. In this study, we identified the transcriptional profiles of bacterial wilt-resistant (ac Yanyan97) and -susceptible (ac Honghuadajinyuan) tobacco cultivars infected with R. solanacearum at six time points by RNA sequencing. Gene expression analysis showed that the resistant cultivar manifested a faster change in the expression of defense-related genes than the susceptible cultivar during R. solanacearum infection, by which more differentially expressed genes (DEGs) were up-regulated rather than down-regulated at all time points. Functional analysis indicated that DEGs were involved in plant hormones, glutathione and secondary metabolic pathways associated with tobacco resistance to bacterial wilt induced by R. solanacearum. Through subsequent Short Time-series Expression Miner (STEM) and weighted correlation network (WGCNA) analyses, the phenylpropanoid metabolic pathway was identified as a key pathway for tobacco defense against R. solanacearum infestation. In summary, our results provide transcriptomic profiles of tobacco response to R. solanacearum infestation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over hundreds of millions of years of evolution, plants have developed a complex defense network against pathogens/insects attack (Dodds and Rathjen 2010). Physical barriers (e.g., wax, cuticle, and lignin) can protect plant cells against complex environmental elements (Underwood 2012). In addition, plants produce some killing weapons, such as secondary metabolites (e.g., alkaloids and phenols) and inhibitory substances (Kuc 1995). Hormones also act as messengers in this battle, which mobilize the expression of related kinase cascade, defense-related genes, transcription factors, and so on (Dermastia 2019; Fesenko et al. 2019; Zhang et al. 2019). Although the effects of hormones are not as rapid as those of turgor changes and calcium oscillations changes, they are undoubtedly a kind of effective and well-known long-distance transport molecules that induce plants to invest more troops and promote a coordinated defense strategy (David et al. 2013; Katagiri 2004; Pieterse et al. 2009). The evolution of these powerful defense systems in plants has enabled them to compete effectively with the evolution of pathogens (Zhang et al. 2019; Anderson et al. 2010; Han 2019).
Tobacco (Nicotiana tabacum) is a widely grown cash crop in the world (Sierro et al. 2014). The total tax revenue of tobacco in China reached 175.2 billion USD in 2017. It has been widely used in applied science and has great scientific value. Tobacco is a versatile model organism for functional genomics and biotechnology applications and is one of the most effective plant material used in expressing foreign proteins (Nagata et al. 1992, Häkkinen et al. 2007; Ameye et al. 2017; Romeis et al. 2001). However, tobacco bacterial wilt caused by R. solanacearum is a devastating soilborne disease worldwide resulting in 10–30% losses in tobacco production (Elphinstone 2005). Therefore, it is important to study the tobacco defense mechanism and uncover the responsive defense genes during R. solanacearum infection.
R. solanacearum has a wide host range, attacking more than 250 plant species among 54 monocot and dicot botanical families, and can survive for long periods under a wide range of environmental conditions (Mansfield et al. 2012). It is a highly diversified pathogen mainly encompassing four evolutionary types, five races and six biovars (Fegan and Prior 2005). Only evolutionary type I physiological race 1 infects tobacco.
The molecular mechanism of the resistant and susceptible tobacco varieties can be divided into two types: qualitative and quantitative. Tobacco plants have the capability to identify several effectors of R. solanacearum (e.g., ripB) which are recognized by the resistance proteins (e.g., Roq1) (Nakano and Mukaihara 2019; Thomas et al. 2020). However, tobacco molecular mechanism underlying quantitative (polygenic) resistance to this important pathogen remains unclear (Peeters et al. 2013). For example, the defense-related genes controlling the bacterial wilt in tobacco, the action and the interaction mechanism between tobacco and R. solanacearum are still indefinite. Therefore, investigation and excavation of possible defense-related genes in tobacco germplasms based on transcriptomic level will not only deepen the understanding of the interaction between tobacco and R. solanacearum, but also will help to make rational use of bacterial wilt defense-related genes, thus serving tobacco breeding programs for disease resistance.
Some studies have reported plant interaction with R. solanacearum, focusing on specific species such as Arabidopsis, tomato and potato. In Arabidopsis, Hu et al. inoculated the roots of the susceptible Col-5 and RRS1-R-containing resistant Nd-1 plants with R. solanacearum strains harboring or lacking the virulence gene, PopP2 (Hu et al. 2008). It was found that ABA, aging and basal defense-related genes were up-regulated in response to disease infection. Inactivation of some disease-associated genes causes changes in the response of plants to pathogenic strains (Hu et al. 2008). Zuluaga et al. used the Scommersonii to infect the roots of the potato resistant accession (F118) and susceptible accession (F97) (Zuluaga et al. 2015). It was found that 22.6% and 12.8% differentially expressed genes were identified as responsive to biotic stress in F118 and F97, respectively, and half of those genes were up-regulated and involved in plants response to pathogens in both cultivars. Hormone-related genes indicated that both ET and JA were induced in the susceptible cultivar F97, but not in resistant cultivar F118. In contrast, SA-related genes were down-regulated in both cultivars after pathogen infection. Similarly, hormone-related genes were highly induced in potato, indicating a significant difference in the response of the two accessions to the pathogens (Narancio et al. 2013). Wang et al. conducted transcriptional and protein-level studies using anti-inductive materials and found that the biosynthesis of methionine cycle (MTC) and down-regulation of γ-aminobutyric acid (GABA) played an important role in the interaction between R. solanacearum and tomato plants (Wang et al. 2019). However, due to the specie specificity, response mechanisms to pathogen infection might be different among the different species. In tobacco, the molecular mechanism of plant resistance is controlled by multiple quantitative trait loci (QTL) which makes it more complex than other model plant species like Arabidopsis (Laurent et al. 2002; Laurence et al. 2010). With regard to tobacco resistance to bacterial wilt, current studies mainly focus on gene mapping, molecular marker-assisted breeding, genetic transformation of defense-related genes and proteomics research. RNA sequencing (RNA-seq) is a favored high-throughput technology for genome-wide gene expression quantification providing highly accurate, and comprehensive transcriptome profiling (Wang et al. 2009). During multi-stages of pathogen infection, genes are differentially expressed, thus studying plant responses in different time points is important. To determine the statistically significant time-dependent gene expression profiles, Short Time-Series Expression Miner (STEM) analysis is an ideal approach to identify genes with similar time-series characteristics (Ernst and Bar-Joseph 2006). Moreover, weighted gene co-expression network analysis (WGCNA) is a novel gene co-expression network-based method that illustrates the molecular interaction mechanism and the relationship between gene networks (Langfelder and Horvath 2008).
The tobacco cultivar 'ac Yanyan97' is a moderately resistant cultivar to tobacco-virulent R. solanacearum strains (Qian et al. 2013; Lan et al. 2014). Here, we studied the response of the ac Yanyan97 (resistant) cultivar and the susceptible cultivar (ac Honghuadajinyuan) to R. solanacearum infection and investigated the potential genes involved in plant defense at different time points. We used gene enrichment and co-expression analyses to explore the role of tobacco DEGs in modeling phytohormones signaling, and focused on phenylpropanoid biosynthesis pathway. This study provides a large-scale transcriptomic dataset that can open new insights into tobacco disease resistance to R. solanacearum.
Materials and methods
Plant materials and inoculation treatments
The N. tabacum cv ac Yanyan97 (Y) resistant cultivar and ac Honghuadajinyuan (H) susceptible cultivar were used in the study. To maximize the purification of the external environment, and avoid erroneous effects on the results, all tobacco plants were grown in 1/2 MS medium (maintaining a sterile and nutrient-rich environment) under 24–26 °C in the daytime and 20–22 °C in the night, under a 16/8 h light/dark photoperiod, until the tobacco plants reached the six-leaf stage (about 45 days after germination). In such a harsh environment, all the plants we obtained for the experiment were similar in terms of tobacco development and health.
For inoculation treatments, the plants were first transferred to a lamina. Then we used a 25 g × 5/8-inch needle syringe to inject 1 cm from the base of the seedling stem (Fig S7-D) 1 mL of OD600 = 1.0 bacterial solution of R. solanacearum LC3-6 strain. LC3-6 of R. Solanacearum was isolated from the diseased plant of Flue-cured tobacco variety Yunyan87 in Baiyangba Town, Lichuan City, Enshi Prefecture, the main tobacco-producing area of Hubei Province, China. LC3-6 is the serial number of the isolated strain identified by Hubei Academy of Tobacco Sciences and Huazhong Agricultural University that belongs to the physiological race 1, biochemical type III, evolutionary type I, sequence variant 17.
The control was treated with sterile water instead of bacterial solution. Samples were collected from 1 cm beneath the crown area including the upper part of the root. The samples were harvested at 0, 12, 24, 36, 48, 72 and 120 HPI (hour post-infection) from three independent biological replicates. All samples were immediately frozen in liquid nitrogen after sampling and stored at − 80 °C.
RNA isolation and quantification
RNeasy plant mini kit was used along with DNase treatment to extract the RNA of plant sample, according to the manufacturer’s instruction (QIAGEN, Germany). RNA Nano 6000 Assay Kit of the Bio analyzer 2100 system (Agilent Technologies, CA, USA) was used to determine the quality and quantity of RNA.
RNA library preparation for transcriptomic analysis
Subsequently, 3 μg total RNA of each sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Second-strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3′ ends of DNA fragments, NEBNext Adaptors with hairpin loop structure were ligated to prepare for hybridization. To preferentially select cDNA fragments of preferentially 250–300 bp in length, the library fragments were purified using the AMPure XP system (Beckman Coulter, Beverly, USA). Then, 3 µL USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min, followed by 5 min at 95 °C before PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index (X) Primer. Finally, the PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system.
Sequencing, assembly, and functional annotation
The clustering of the index-coded samples was performed on the cBot Cluster Generation System using the TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq platform and paired-end reads of 125/150 bp were generated. In this step, clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N and low-quality reads from raw data by fastqc. At the same time, the clean data for Q20, Q30 and GC content were calculated. All the downstream analyses were based on the clean data with high quality. Reference genome, N. tabacum K326 genome (Sierro et al. 2014), and gene model annotation files were downloaded directly from SGN website. The index of the reference genome was built using Hisat2 and the paired-end clean reads were aligned to the reference genome using Hisat2 (v2.0.5) (Sierro et al. 2014; Daehwan et al. 2015). We selected Hisat2 as the mapping tool for that Hisat2 can generate a database of splice junctions based on the gene model annotation file, which obtain a better mapping result than other non-splice mapping tools (Mortazavi et al. 2008).
cDNA preparation and qRT-PCR
Candidate transcriptome genes were verified by qRT-PCR analysis using three biological replicates. Gene-specific primers were designed using Primer 5.0 and commercially synthesized (Qinco, China). The first-strand cDNA was synthesized from 2 μg of total RNA using SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer's instructions and reverse transcribed into cDNA, followed by 100-fold dilution. RT-PCR was performed in a 10 μL reaction volume containing 5 μL of 100 × diluted cDNA as a template, each with 0.25 μL of forward and reverse gene-specific primers, and 4.5 μL of SsoFast EvaGreen Supermix with low ROX (Bio-Rad, Hercules, CA). The qRT-PCR reaction was carried out on an Applied Biosystem 7500 Real-Time PCR System (Applied Biosystem, Foster City, CA) at 95 °C for 2 min, then at 95 °C for 40 cycles of 5 s and at 60 °C for 35 s. The qRT-PCR product ranged from 80 to 200 bp. The relative quantification of gene expression was calculated and normalized using EF-1α (Accession No: LOC107788982) as an internal standard. After qRT-PCR, the dissolution profile was used to confirm the specificity of the primers. The dissociation parameters were 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 15 s. The Ct(2−ΔΔCt) method was compared to calculate the fold change in the gene expression levels (Pfaffl 2001).
Gene network construction and visualization (WCGNA)
Co-expression networks were constructed using the WGCNA (v1.29) package in R. We drew heatmaps for each module based on correlation coefficients, with a deeper color representing a higher degree of correlation. To further explore interactions among genes in each module, we selected those genes with the highest connectivity to draw the gene network. In addition, information regarding the functions of differentially expressed genes was collected from Unigene annotations, and these genes were subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Ogata et al. 2000) significant enrichment analyses to identify the biological functions and metabolic pathways in which these genes participate. Cytoscape (Shannon et al. 2003) was used to visualize the final interaction network.
Short time-series expression miner (STEM) analysis
The STEM analysis of the total DEGs of four important temporal gene expression profiles (81_Y, 20_Y, 81_H and 20_H) was performed using the STEM version 1.3.11 developed by (Ernst and Bar-Joseph 2006). The studied DEGs were all stratified into different profiles based upon various expression patterns calculated by STEM analysis, respectively.
Results
Global transcriptome profiling of tobacco response to R. solanacearum infection
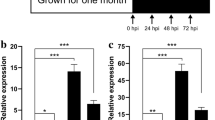
A total of ~ 2.58 billion raw reads were obtained from 78 libraries, and 25–52 million reads were generated by each library. In total, ~ 767G clean bases were obtained and ~ 0.3% of the total pair-end reads were filtered and trimmed. The transcriptome data was aligned with the N. tabacum cv. K326 reference genome (Sierro et al. 2014) and approximately ~ 92% of the clean reads were mapped to ~ 88,000 transcripts. Afterward, we performed a principal component analysis (PCA) with normalized read counts to assess the overall differences of the infected transcriptome at different time points and provided an overview of the transcriptome landscape in the different cultivars and time points (Fig. 1A). The resistant (Y3, Y5, Y7, Y9, Y11, Y13) and susceptible (H3, H5, H7, H9, H11, H13) cultivars can be obviously distinguished from each other, and the control groups were obviously clustered together (Y1, Y2, Y4, Y6, Y8, Y10, Y12) and (H1, H2, H4, H6, H8, H10, H12), respectively. The resistant cultivar is more clearly separated in the 24 HPI treatment, while the susceptible cultivar is more distinct until 48 HPI. Samples at 12, 24, 36, 48, 72 and 120 HPI were clustered far away from control samples, which show that R. solanacearum infection induced different gene expression profiles. To analyze the expression level of all genes, we used the total mapped reads of all genes, calculating gene expression level as fragments per kilobase of exon model per million mapped fragments (FPKM) (Figs. S1 and S2). Genes with false discovery rate (FDR) < 0.05 and absolute value of log2 (FC) ≥ 2 were selected as differentially expressed genes (DEGs) for further analysis. When cross-comparing RNA-seq data, we detected a total of ~ 27,818 (~ 25%) DEGs between the resistant and susceptible cultivars at least in one time point (Table S1). Compared with the mock group (water treatment), more up-regulated genes were detected in the Y and H cultivars than down-regulated genes at the early time points of inoculation. Y cultivar at 12, 24, 36, 48, 72 and 120 h exhibited 612 (452 up and 160 down), 5067 (3252 up and 1815 down), 5406 (3601 up and 1805 down), 8606 (5216 up and 3390 down), 10,148 (5582 up and 4566 down) and 8464 (5909 up and 2555 down) DEGs, respectively. The H samples at 12, 24, 36, 48, 72 and 120 h exhibited 288 (144 up and 44 down), 322 (246 up and 76 down), 964 (883 up and 81 down), 3504 (2391 up and 1113 down), 5376 (3489 up and 1887 down) and 13,912 (7581 up and 6331 down) DEGs, respectively (Fig. 1B, Tables S2 and S3). The number of DEGs in the resistant cultivar was significantly higher than that in the susceptible cultivar at 120 h, and the number of DEGs in the resistant cultivar increased significantly after 24 h of infection. Interestingly, the number of DEGs in the Y cultivar decreased after 120 h of inoculation, while the number of DEGs of the susceptible cultivar from the beginning to end showed an upward trend.

Landscape of transcriptomics in tobacco response to R. solanacearum at six time points. a PCA plot for RNA-seq dataset at 12, 24, 36, 48, 72 and 120 h time points after R. solanacearum inoculation in resistant (Y) and susceptible (H) tobacco cultivars. b Number of genes significantly changed in the expression of Y and H cultivars following R. solanacearum infection. Fold change in gene expression was calculated as log2 (log2FC) in tobacco with and without R. solanacearum infection at the same time points. c Venn diagrams illustrating the number of up- and down-regulated transcripts over the time course of R. solanacearum infection
The distribution of up- and down-regulated DEGs at the six time points after inoculation was analyzed. A total of 24 DEGs, 396 DEGs and 1676 DEGs were detected only at 12 vs 24 h, 36 vs 48 h and 72 vs 120 h in both Y and H after inoculation, respectively (Fig. 1C). We found that the number of shared DEGs in the Y and H cultivars showed an increasing trend at the late stage of inoculation. These results indicate that the changes in gene expression of Y and H cultivars had similarities, and their resistant mechanisms had commonalities. The resistant Y cultivar had more differentially expressed genes that differed and responded earlier to the bacterial infection. However, fewer number of constitutive DEGs among all time points were expressed in the H group (Fig. S3).
Functional analysis of DEGs in resistant and susceptible tobacco cultivars under R. solanacearum infection
To elucidate the involved primary metabolic pathways, the DEGs at each time point were subjected to KEGG pathway analysis (Table S2). Compared with the sterile water control, the DEGs at 12, 24, 36, 48, 72 and 120 h were significantly assigned to 14, 10, 8, 5, 22 and 13 pathways in the Y cultivar, as well as 14, 14, 13, 22, 17, and 21 pathways in the H cultivar (P < 0.05), respectively. The top ten pathways of each time point are listed in Fig. 2. Of these significant pathways, glutathione metabolism, phenylpropanoid biosynthesis, cysteine and methionine metabolism, oxidative phosphorylation, MAPK signaling pathway, plant–pathogen interaction and plant hormone signal transduction were involved in the response of the Y cultivar to R. solanacearum at all post-infestation time points (Fig. 2A). Similarly, glutathione metabolism, phenylpropanoid biosynthesis, alpha-linolenic acid metabolism, and arginine and proline metabolism were involved in the response of the H cultivar to R. solanacearum at all post-infestation time points (Fig. 2B). KEGG results showed that the phenylpropanoid biosynthesis and glutathione metabolism pathways were enriched in both resistant and susceptible tobacco.

KEGG pathway enrichment analysis of DEGs in the tobacco transcriptome induced by R. solanacearum in six time points. Data were visualized using a scatter diagram with q-value levels indicated by ‘ − log10 (padj)’ and an enrichment factor indicative of individual pathways. Values in parentheses represent the number of components in each pathway present in the DEG dataset. The right panel represents KEGG pathway combinations at different time points (parentheses number) in the Y (a) and H (b) cultivars
Dynamic differences identification in tobacco transcriptome through short time-series expression miner (STEM) analysis
To understand the dynamic differences of tobacco transcriptome in response to R. solanacearum infection, we carried out Short Time-series Expression Miner (STEM) analysis with total DEGs (Ernst and Bar-Joseph 2006). Four important temporal gene expression profiles (81_Y, 20_Y, 81_H and 20_H) are presented in the plates (Fig. 3, Table S3). Profiles 81_Y and 20_Y contained genes from the Y cultivar within 12–120 HPI following the onset of R. solanacearum infection. Profile 81_Y embodied the most transcripts (2764), in which DEGs of this profile controlled the glutathione metabolism, plant–pathogen interaction and MAPK signaling pathway (Fig. 3A). These transcripts were up-regulated immediately after 12 h of infection and this up-regulation continued in the same trend until the latest time of infection 120 h, suggesting that the invasion of R. solanacearum induced these defense pathways continuously. Most of the genes in the 20_Y profile were involved in the primary metabolism of carbohydrates, lipids and amino acids.


Time-series transcriptome analysis of significant DEGs induced by R. solanacearum infection in tobacco. Example of cluster81 and cluster20 in Y (a) and H (b) tobacco cultivar infection R. solanacearum. The top 20 KEGG pathways for each profile are listed in the lower portion. Enrichment scores are shown as -log10 (padj)
The 81_H and 20_H profiles contained genes from H cultivar within 12–120 HPI following onset of R. solanacearum infection. The (1,433) up-regulated genes of profile 81 were mainly involved in some primary metabolism pathways (e.g., glutathione metabolism, nucleotides, carbohydrates and lipids, as well as energy production) (Fig. 3B). In the 81_H profile, genes controlling the circadian rhythm were significantly up-regulated only at 24 h after R. solanacearum infection, while the phenylpropanoid biosynthesis pathway was within the down-regulated genes of the 20_H profile. For the phenylpropanoid metabolic pathway, there is a significant difference between the Y and H materials; in the susceptible material H, it always shows a downward trend, while in the disease-resistant material Y, there are two small up-regulated crests.
Co-expression network construction of tobacco plants under R. solanacearum infection
To identify the activation of specific biological processes, the potential relationships between modules (co-expression genes) and traits need to be determined. Therefore, based on the log2 (FPKM) values, we constructed a weighted gene co-expression network using a soft thresholding power (β = 6) with higher adjacency (Fig. S4). Using a heatmap-based plot incorporating different module assignments and gene dendrograms, we initially visualized the topological overlap matrix (TOM) of DEGs after R. solanacearum infestation (Fig. 4A). Obviously, each module in this chart had high overlaps, so we divided the network into 19 modules. Then we performed gene expression pattern plots on these 19 modules to analyze the expression characteristics of each module. As shown in Fig. 4B, the expression was characterized by obvious up-regulated expression after inoculation. The gene KEGG enrichment analysis of light red module was found to be significantly enriched in phenylpropane, phenylalanine and flavonoid metabolic pathways (Fig. 4C). Further graphics presented some valuable genes related to flavonoid metabolic pathways, such as C4H, 4CL, and peroxidase that served as hub genes (Fig. 4D). They appear to be central genes playing significant role in plant–pathogen interactions. The interactions trigger the regulation of plant primary metabolic genes.


Weighted gene co-expression network analysis. a Heatmap of the gene network light colors represent low overlap, and gradually darkened red represents higher overlap of gene pairs from various data sets. Diagonal lines represent different modules. b An example of one module heatmaps in each network module. c KEGG pathway enrichment analysis of the light yellow module. d The most highly connected gene network in the DEGs light-yellow module after R. solanacearum infection. The red node represents the anti-correlation gene, while the blue node represents the gene of unknown function
Potential genes involved in tobacco resistant to R. solanacearum
Through the previous analysis, phenylpropanoids pathway occurred at all time points in the Y and H cultivars (Figs. 2, 3, 4C). The phenylpropanoid metabolic pathway contains numerous important precursors involving the biosynthesis of peroxidase and secondary metabolites that are closely related to plant disease resistance. On further analysis, we identified a diverse group of defense-related genes that have differentially transcriptional responses after R. solanacearum infestation. The majority of these genes belong to metabolite synthase, hormone and pathogen-related genes. The gene families that could be interesting targets for disease resistance engineering are further elaborated below.
To confirm the RNA-seq results, the expression profiles of several selected DEGs were tested by quantitative real-time PCR (qRT-PCR). The high correlation coefficients of log2-fold changes obtained from RNA-seq and qRT-PCR results suggested that the RNA-seq data in this study were validated (Fig. S5).
Oxidative stress-related genes induced by R. solanacearum attack
Reactive oxygen species (ROS) play an important role in plant responses to biotic stresses including pathogenic bacteria infestation. Interestingly, we found that the response of peroxidase genes was different before and after infection. The expression of several peroxidase genes was significantly up-regulated at 24 h after inoculation in the Y material, while the susceptible material (H) showed normal expression (Fig. 5). This result is consistent with other reports which indicated that the peroxidase gene is involved in the plant defense system against R. solanacearum (Brown and Allen 2004; Flores-Cruz and Allen 2009).

Heatmap of phenylpropanoid biosynthesis-related gene expression. Values are presented as fold change relative to control (water treatment in the same period)
Secondary metabolites-related genes induced by R. solanacearum attack
Low molecular weight secondary metabolites (called phytoalexins) are one of the best defensive tools against pathogen infection in plants. Therefore, we focused on the enriched DEGs of secondary metabolites in the phenylpropanoid metabolic pathway. Interestingly, we found that genes related to scopoletin synthesis were up-regulated in the Y cultivar (Fig. 6), which was more obvious in the mid-stage of inoculation, while nicotine synthesis-related genes were up-regulated more obviously in the H cultivar, especially in the early stage of inoculation. From this point of view, it seems that the antibacterial effect of scopoletin induced resistance to R. solanacearum infection. Some studies have found that the plant resistance to bacterial pathogens is closely related to the rate and intensity of the accumulation of scopoletin (Goy et al. 1993), which is in agreement with the results found in this study.

Effects of R. solanacearum infestation on secondary metabolites biosynthesis. Heatmap of secondary metabolite biosynthesis-related gene expression. Values are presented as fold change relative to the control (the same period of water treatment)
Plant hormone-related genes induced by R. solanacearum attack
Previous studies have found that crop plants showed effective resistance against R. solanacearum infection through the regulation of hormone. The primary role of salicylic acid (SA) is to establish systemic acquired resistance (SAR), a long-lasting and broad-spectrum form of disease resistance (David et al. 2013; Durrant and Dong 2004). To determine the changes of SA-related genes in tobacco plants after R. solanacearum infection, we analyzed the log2fold-changes at 12, 24, 36, 48, 72 and 120 h Fig. S6. Results revealed a significant increase in the synthesis-related genes of SA in both cultivars infected by R. solanacearum; however the increase in the disease-resistant cultivar (Y) was more obvious (Fig. 7). Isochorismate synthase (ICS) and phenylalanine ammonia lyase (PAL) are the two major genes involved in ICS and PAL synthetic pathways, respectively, which together participate to synthesize PR1, which is also a major gene for SA signal transduction (Dempsey et al. 2011). Following R. solanacearum infestation, all PAL, ICS and PR1 genes were up-regulated in the early stage of infection, and the expression trend in Y materials was obviously high.

Effects of R. solanacearum attacking tobacco plants on hormone biosynthesis-related genes. This is a heatmap of SA biosynthesis-related gene expression. Values are presented as fold change relative to mock
Hereafter, we analyzed other hormone signaling pathways. The auxin signaling-related genes were up-regulated in the early inoculation stage, but the up-regulation trend was more obvious in the susceptible materials. Genes related to jasmonic acid (JA) and ethylene signaling pathways showed also up-regulated expression and the trend of the up-regulation in the disease-resistant materials was more obvious. These results indicate that these genes might have effective defense effects, which deserve further exploration.
Discussion
Tobacco bacterial wilt is one of the most economically important soilborne diseases that affects significantly the yield and quality of tobacco. R. solanacearum can survive in non-host plants or soil; once the host plant appears, it causes a devastating damage (Genin 2010; Stéphane 2010). Plant response to a pathogenic bacterium is a complex process which depends on many factors. Up to now, there is no in-depth understanding of a series of reactions induced by R. solanacearum in tobacco (Peeters et al. 2013). The transcriptional response of plants to pathogen infection is a dynamic and complex reprogramming process composed of signals and compounds, which varies with the time of infection (Libault 2018; Fendrych et al. 2018; Kong et al. 2017). Studies on the transcriptome response of tobacco to R. solanacearum infection are rare, mainly focused on small genomes such as Arabidopsis, tomato and potato using leaf samples and untreated materials as control. Moreover, the studying period is short, so the results obtained cannot accurately reflect the changes in the transcriptome level during R. solanacearum infection.
In this study, we analyzed the dynamic changes of the transcriptome profiles of two tobacco cultivars showing different resistance level to R. solanacearum infection as shown in Fig S7. Unlike previous studies, we used tissue culture seedlings (sterile seedlings) as experimental materials to avoid the possible interference of the external environment, especially the complex rhizosphere environment, on the transcriptome results. At the same time, the actual production experience shows that the grafted material improves resistance to R. solanacearum, so RNA-seq samples were collected from 1 cm beneath the crown area including the upper roots. Controlled by R. solanacearum-specific variables, our method ensured further specificity by the purified environment and analyzed the transcriptome profiles regarding only plant responses to the bacterial infection.
Through the KEGG enrichment analysis of disease-resistant (Y) and -susceptible materials (H) at different time points, it was found that the two materials had the same enrichment results including phenylpropanoid biosynthesis, glutathione metabolism and other pathways (Fig. 2). To further judge whether these pathways differed in the two materials, we performed an STEM analysis and found that glutathione metabolism showed up-regulated trend, while phenylpropanoid displayed down-regulated trend in the susceptible materials (Fig. 3). This might explain the different tolerance level between the two materials which was further verified by WGCNA analysis (Fig. 4).
Previous analysis showed that the phenylpropanoid pathway frequently occurred (Figs. 2, 3, 4C). The phenylpropanoid metabolic pathway contains numerous disease resistance precursors of secondary metabolites and salicylic acid synthesis. Salicylic acid is one of the key defense-related hormones that activates defense responses against biological and semi-biotrophic pathogens (Loake and Grant 2007). The primary role of SA is to establish SAR (Durrant and Dong 2004). Previously, it was reported that mutants of NahG gene, which can degrade SA in plants, were less resistant to R. solanacearum indicating positive correlation between SA and plant resistance to bacterial wilt (Denance et al. 2013). Interestingly, our study found that the expression of ET-, ABA-, JA-, and SA-related genes was up-regulated in both cultivars, and all of these differentially expressed genes were relatively more pronounced in the resistant cultivar (Fig. 7).
Pathogens can enhance host susceptibility by manipulating hormone signaling (Dermastia 2019, Katagiri 2004; David et al. 2013, Zhang et al. 2019). Similarly, the response of tobacco plants to R. solanacearum infection is also in line with this characteristic, showing a close correlation with various hormones. The comparative proteome studies of resistant cultivars showed that the expression of apical membrane antigen might enhance the resistance of tomato to R. solanacearum, while the apical membrane antigen was regulated by SA. In comparison of tomato resistant cultivars, it was found that the expression of signal transduction-related genes of ET, JA, and IAA was up-regulated, and the expression of GA-related genes was down-regulated (Takeaki et al. 2012). Moreover, in potato resistant cultivars, the expression of ET and JA signaling-related genes was up-regulated, while the expression of SA-related genes was down-regulated in both ecotypes (Zuluaga et al. 2015). Interestingly, our study found that the expression of ET-, ABA-, JA-, and SA-related genes was up-regulated in both cultivars, and all of these differentially expressed genes were relatively more pronounced in the resistant cultivars (Fig. 7).
Low molecular weight secondary metabolites (called phytoalexins) are one of the best plant defense means against pathogen infection (Kuc 1995; Hammerschmidt 1999; Ahuja et al. 2012). Since the introduction of the phytoalexin hypothesis in 1940, the defense effects of these compounds have been proven effectively by various experimental methods (Hammerschmidt 1999). The phenylpropanoid metabolic pathway is a precursor for the synthesis of many metabolites, such as nicotine, scopoletin and so on, which are often closely related to plant resistance (Sun et al. 2014; Seo et al. 2012). Based on our RNA-seq data, the expression of genes related to scopoletin synthesis was up-regulated in the resistant cultivar, especially in the mid-stage of inoculation, while the expression of genes related to nicotine synthesis was up-regulated in the susceptible cultivars, especially at the early stage of inoculation. From this point of view, it seems that scopoletin is more effective in dealing with the infection of R. solanacearum (Fig. 6). This is in line with its reported role as an antibacterial compound, where the level of scopoletin significantly increases after pathogen invasion and exhibits pathogenic toxicity in vitro (Carpinella et al. 2005; El-Oirdi et al. 2010; Silva et al. 2002; Gnonlonfin et al. 2012). The resistance of some plant species to pathogens was linked to the rate and intensity of scopoletin accumulation (Gnonlonfin et al. 2012). These results represent a very important step in assessing the defense function of the scopoletin.
In addition, peroxidase genes are associated with plant defense against R. solanacearum. Interestingly, we found that peroxidase responsive genes expressed differently before and after R. solanacearum infection (Fig. 5). The Y martials reveal up-regulated transcript of peroxidase responsive genes, the highest transcript was at 24 HPI. The H martials also showed synchronic increase in the level of peroxidase responsive genes with the increase of the time of infection, but not as high as that of Y martials.
To sum up, we studied the direct and indirect defense responses of two tobacco cultivars, ac Yanyan97 (resistant) and ac Honghuadajinyuan (susceptible), grown under aseptic conditions to R. solanacearum infection at different time points (Table 1). Dynamic transcriptome analysis showed a strong transcriptomic response 48 h after infection that continued until 120 HPI. During these stages, the phytohormones scopoletin and peroxidase were involved in tobacco resistance to the bacterial wilt. This work reveals the interaction between tobacco and R. solanacearum for the first time and not only provides some insights into the molecular and biochemical mechanisms of tobacco resistance to R. solanacearum, but also highlights the uniqueness of tobacco itself. The parallelly expressed defensive genes reported in this study may be of great significance and could contribute to the progress of tobacco breeding for bacterial wilt resistance.
References
Ahuja I, Kissen R, Bones AM (2012) Phytoalexins in defense against pathogens. Trends Plant Sci 2:73–90
Ameye M, Allmann S, Verwaeren J, Smagghe G, Haesaer G, Schuurink RC, Audenaert K (2017) Green leaf volatile production by plants: a meta-analysis. New Phyto 3:666–683
Anderson JP, Gleason CA, Foley RC, Thrall PH, Burdon JB, Singh KB (2010) Plants versus pathogens: an evolutionary arms race. Funct Plant Biol 6:499–512
Brown DG, Allen C (2004) Ralstonia solanacearum genes induced during growth in tomato: an inside view of bacterial wilt. Mol Microbiol 6:1641–1660
Carpinella MC, Ferrayoli CG, Palacios SM (2005) Antifungal synergistic effect of scopoletin, a hydroxycoumarin isolated from Melia azedarach L. fruits. J Agric Food Chem 8:2922–2927
Daehwan K, Ben L, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4):357–360
David DV, Godelieve G, Monica HF (2013) Hormone defense networking in rice: tales from a different world. Trends Plant Sci 18(10):555–565
Dempsey DA, Vlo AC, Wildermuth MC, Klessig DF (2011) Salicylic acid biosynthesis and metabolism. Arabidopsis Book 9:e0156
Denance N, Ranocha P, Oria N, Barlet X, Riviere MP, Yadeta KA, Hoffmann L, Perreau F, Clement G, Maia-Grondard A, van den Berg GC, Savelli B, Fournier S, Aubert Y, Pelletier S, Thomma BP, Molina A, Jouanin L, Marco Y, Goffner D (2013) Arabidopsis wat1 (walls are thin1)-mediated resistance to the bacterial vascular pathogen, Ralstonia solanacearum, is accompanied by cross-regulation of salicylic acid and tryptophan metabolism. Plant J 2:225–239
Dermastia M (2019) Plant hormones in phytoplasma infected plants. Front Plant Sci 10:477
Dodds PN, Rathjen JP (2010) Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet 8:539–548
Durrant WE, Dong X (2004) Systemic acquired resistance. Annl Rev Phytopathol 42:185–209
El-Oirdi M, Trapani A, Bouarab K (2010) The nature of tobacco resistance against Botrytis cinerea depends on the infection structures of the pathogen. Environ Microbiol 1:239–253
Elphinstone J (2005) The current bacterial wilt situation: a global overview. Bacterial wilt disease and the Ralstonia solanacearum species complex. APS press, Minnesota, USA, pp 9–28
Ernst J, Bar-Joseph Z (2006) STEM: a tool for the analysis of short time series gene expression data. BMC Bioinform 7:191
Fegan M, Prior P (2005) How complex is the Ralstonia solanacearum species complex. In: Allen C, Prior P, Hayward AC (eds) Bacterial wilt disease and the Ralstonia solanacearum species complex. APS Press, St Paul, MN, pp 449–461
Fendrych M, Akhmanova M, Merrin J, Glanc M, Hagihara S, Takahashi K, Uchida N, Torii KU, Friml J (2018) Rapid and reversible root growth inhibition by TIR1 auxin signalling. Nat Plants 7:453–459
Fesenko I, Azarkina R, Kirov I, Kniazev A, Filippova A, Grafskaia E, Lazarev V, Zgoda V, Butenko I, Bukato O, Lyapina I, Nazarenko D, Elansky S, Mamaeva A, Ivanov V, Govorun V (2019) Phytohormone treatment induces generation of cryptic peptides with antimicrobial activity in the moss Physcomitrella patens. BMC Plant Biol 1:9
Flores-Cruz Z, Allen C (2009) Ralstonia solanacearum encounters an oxidative environment during tomato infection. Mol Plant Microbe Interact 7:773–782
Genin S (2010) Molecular traits controlling host range and adaptation to plants in Ralstonia solanacearum. New Phytol 4:920–928
Gnonlonfin GJB, Sanni A, Brimer L (2012) Review scopoletin—a coumarin phytoalexin with medicinal properties. Crit Rev Plant Sci 1:47–56
Goy PA, Signer H, Reist R, Aichholz R, Blum W, Schmidt E, Kessman H (1993) Accumulation of scopoletin is associated with the high disease resistance of the hybrid Nicotiana glutinosa x Nicotiana debneyi. Planta 191:200–206
Häkkinen ST, Tilleman S, Šwiątek A, Sutter VD, Rischer H, Vanhoutte I, Onckelen HV, Hilson P, Inzé D, Oksman-Caldentey KM (2007) Functional characterisation of genes involved in pyridine alkaloid biosynthesis in tobacco. Phytochem 22:2773–2785
Hammerschmidt R (1999) Phytoalexins: what have we learned after 60 years? Ann Rev Phytopathol 37:285–306
Han GZ (2019) Origin and evolution of the plant immune system. New Phytol 1:70–83
Hu J, Barlet X, Deslandes L, Hirsch J, Feng DX, Somssich I, Marco Y (2008) Transcriptional responses of Arabidopsis thaliana during wilt disease caused by the soil-borne phytopathogenic bacterium, Ralstonia solanacearum. PLoS One 3(7):e2589
Katagiri F (2004) A global view of defense gene expression regulation—a highly interconnected signaling network. Curr Opin Plant Biol 5:506–511
Kong L, Qiu X, Kang J, Wang Y, Chen H, Huang J, Qiu M, Zhao Y, Kong G, Ma Z, Wang Y, Ye W, Dong S, Ma W, Wang Y (2017) A phytophthora effector manipulates host histone acetylation and reprograms defense gene expression to promote infection. Curr Biol 7:981–991
Kuc J (1995) Phytoalexins, stress metabolism, and disease resistance in plant. Ann Rev Phytopathol 1:275–297
Lan T, Zheng SF, Yang L, Wu SX, Wang B, Zhang SJ, Tong ZJ, Chen YZ, Chen SH, Duan YL, Wu WR (2014) Mapping of quantitative trait loci conferring resistance to bacterial wilt in tobacco (Nicotiana tabacum L.). Plant Breed 113:672–677
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform 9:559
Laurence G, Laurent S, Torii KU, Olivier G, Brigitte M, Grimsley NH, Marco Y (2010) ERECTA, an LRR receptor-like kinase protein controlling development pleiotropically affects resistance to bacterial wilt. Plant J 36(3):353–365
Laurent D, Jocelyne O, Frederic T, Judith H, Xin FD, Peter BE, Jim B, Marco Y (2002) Resistance to Ralstonia solanacearum in Arabidopsis thaliana is conferred by the recessive RRS1-R gene, a member of a novel family of resistance genes. Proc Natl Acad Sci USA 4:2404–2409
Libault M (2018) Transcriptional reprogramming of legume genomes: perspective and challenges associated with single-cell and single cell-type approaches during nodule development. Front Plant Sci 9:1600
Loake G, Grant M (2007) Salicylic acid in plant defence—the players and protagonists. Curr Opin Plant Biol 5:466–472
Mansfield J, Genin S, Magori S, Citovsky V, Sriariyanum M, Ronald P, Dow M, Verdier V, Beer SV, Machado MA, Toth I, Salmond G, Foster GD (2012) Top 10 plant pathogenic bacteria in molecular plant pathology. Mol Plant Pathol 6:614–629
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 7:621–628
Nagata T, Nemoto Y, Hasezawa S (1992) Tobacco BY-2 cell line as the “HeLa” cell in the cell biology of higher plants. Int Rev Cytol 8:1–30
Nakano M, Mukaihara T (2019) The type III effector RipB from Ralstonia solanacearum RS1000 acts as a major avirulence factor in Nicotiana benthamiana and other Nicotiana species. Mol Plant Pathol 9:1237–1251
Narancio R, Zorrilla P, Robello C, Gonzalez M, Vilaró F, Pritsch C, Rizza MD (2013) Insights on gene expression response of a characterized resistant genotype of Solanum commersonii Dunagainst Ralstonia solanacearum. Eur J Plant Pathol 4:823–835
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 1:29–34
Peeters N, Guidot A, Vailleau F, Valls M (2013) Ralstonia solanacearum, a widespread bacterial plant pathogen in the post-genomic era. Mol Plant Pathol 7:651–662
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 9:e45
Pieterse CM, Leon-Reyes A, Van der Ent S, Van Wees SC (2009) Networking by small-molecule hormones in plant immunity. Nat Chem Biol 5:308–316
Qian Y, Wang X, Wang D, Zhang LN, Zu LC, Gao ZL, Zhang HJ, Wang ZY, Sun XY, Yao DN (2013) The detection of QTLs controlling bacterial wilt resistance in tobacco (N. tabacum L.). Euphytica 192:259–266
Romeis T, Ludwig AA, Martin R, Jones JD (2001) Calcium-dependent protein kinases play an essential role in a plant defence response. EMBO J 20:5556–5567
Seo S, Gomi K, Kaku H, Abe H, Seto H, Nakatsu S, Neya M, Kobayashi M, Nakaho K, Ichinose Y, Mitsuhara I, Ohashi Y (2012) Identification of natural diterpenes that inhibit bacterial wilt disease in tobacco, tomato and Arabidopsis. Plant Cell Physiol 8:1432–1444
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 11:2498–2504
Sierro N, Battey JN, Ouadi S, Bakaher N, Bovet L, Willig A, Goepfert S, Peitsch MC, Ivanov NV (2014) The tobacco genome sequence and its comparison with those of tomato and potato. Nat Commun 5:3833
Silva WP, Deraniyagala SA, Wijesundera RL, Karunanayake EH, Priyanka UM (2002) Isolation of scopoletin from leaves of Hevea brasiliensis and the effect of scopoletin on pathogens of Hbrasiliensis. Mycopathologia 4:199–202
Stéphane G (2010) Molecular traits controlling host range and adaptation to plants in Ralstonia solanacearum. New Phytol 4:920–928
Sun H, Wang L, Zhang B, Ma J, Hettenhausen C, Cao G, Sun G, Wu J, Wu J (2014) Scopoletin is a phytoalexin against Alternaria alternata in wild tobacco dependent on jasmonate signalling. J Exp Bot 15:4305–4315
Takeaki I, Ichiro M, Hideki T, Nakaho K (2012) Transcriptome analysis of quantitative resistance-specific response upon Ralstonia solanacearum infection in tomato. PLoS ONE 10:e46763
Thomas NC, Hendrich CG, Gill US, Allen C, Hutton SF, Schultink A (2020) The immune receptor Roq1 confers resistance to the bacterial pathogens Xanthomonas, Pseudomonas syringae, and Ralstonia in Tomato. Front Plant Sci 11:463
Underwood W (2012) The plant cell wall: a dynamic barrier against pathogen invasion. Front Plant Sci 3:85
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nature Rev Genet 10(1):57–63
Wang G, Kong J, Cui D, Zhao H, Niu Y, Xu M, Jiang G, Zhao Y, Wang W (2019) Resistance against Ralstonia solanacearum in tomato depends on the methionine cycle and the gamma-aminobutyric acid metabolic pathway. Plant J 6:1032–1047
Zhang R, Zheng F, Wei S, Zhang S, Li G, Cao P, Zhao S (2019) Evolution of disease defense genes and their regulators in plants. Int J Mol Sci 2:335
Zuluaga AP, Sole M, Lu H, Gongora-Castillo E, Vaillancourt B, Coll N, Buell CR, Valls M (2015) Transcriptome responses to Ralstonia solanacearum infection in the roots of the wild potato Solanum commersonii. BMC Genomics 16:246
Funding
This work was supported by the Tobacco Research Institute of Hubei Province (027Y2018-007).
Author information
Authors and Affiliations
Contributions
SJ and JC conceived and designed the research. MA, HW, JC, YS, TW, HZ, YL, and CW performed the experiments and analyzed the dataset. MA and HW wrote the manuscript, which was revised by JC and SJ.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Alariqi, M., Wei, H., Cheng, J. et al. Large-scale comparative transcriptome analysis of Nicotiana tabacum response to Ralstonia solanacearum infection. Plant Biotechnol Rep 16, 757–775 (2022). https://doi.org/10.1007/s11816-022-00765-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-022-00765-x