
Abstract
A DNA extraction method using Chelex 100 is widely used for bacteria, Chlamydomonas, and animal cell lines, but only rarely for plant materials due to the need for additional time-consuming and tedious steps. We have modified the Chelex 100 protocol and successfully developed a rapid and simple method of DNA extraction for efficient PCR-based detection of transgenes from a variety of transgenic plant and algal species. Our protocol consists of homogenizing plant tissue with a pestle, boiling the homogenized tissue in a microfuge tube with 5% Chelex 100 for 5 min, and centrifuging the boiled mixture. The supernatant, which is used for PCR analysis, was able to successfully amplify transgenes in transgenic tobacco, tomato, potato, Arabidopsis, rice, strawberry, Spirodela polyrhiza, Chlamydomonas, and Porphyra tenera. The entire DNA extraction procedure requires <15 min and is therefore comparable to that used for bacteria, Chlamydomonas, and animal cell lines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The PCR is the most frequently used technique for amplification of specific DNA fragments in vitro. Because of its high sensitivity, PCR does not require high-quality template DNA obtained using the complicated conventional method with proteinase K, phenol, and chloroform. Numerous shortcuts have been reported for extracting DNA suitable for PCR amplification; of these, the method using the chelating resin Chelex 100 (Walsh et al. 1991) is most popular for extracting DNA from a vast array of biological materials. In the Chelex method, a boiling step alone is sufficient to extract DNA from Escherichia coli suitable for amplifying a gene of interest using PCR. For animal cells and Chlamydomonas, a rapid and simple DNA extraction protocol using Chelex 100 and/or ethylenediaminetetraacetic acid is widely used. However, a similarly uncomplicated DNA extraction protocol is not yet available for plant materials due to the difficulties in extracting DNA from limited amounts of plant tissue.
Chelex 100 has proven to be efficient in extracting DNA for use in PCR analyses in a wide range of experiments (Walsh et al. 1991; Ward 1992; Holmes et al. 1994; Cao et al. 2009). It also removes PCR inhibitors, such as heme and its metabolic products (Panaccio 1991), acidic polysaccharides (Furukawa and Bahavanandan 1983), mineral ions, and humic acids (Tsai and Olson 1992; Tebbe and Vahjen 1993). However, plant DNA extraction with Chelex 100 requires additional time-consuming and tedious steps, including ethanol treatment and autoclaving (Berthold et al. 1993) and, consequently, this method is seldom used to extract DNA from plant material.
We report here our development of a rapid and simple plant DNA extraction technique using Chelex 100 that produces DNA suitable for PCR amplification. This method requires no ethanol treatment or autoclaving steps and can be completed in <15 min. Using this method, we were able to detect transgenes from a variety of transgenic plant and algal species.
Materials and methods
Plant materials
To evaluate our newly developed rapid DNA extraction method, we first generated transgenic plants of various species. Transgenic tobacco (Nicotiana tabacum cv. Xanthi), tomato (Lycopersicon esculentum cv. Joyful), and potato plants (Solanum tuberosum cv. Desiree) were produced via transforming TBP19 (P19 protein of Tomato bushy stunt virus, GI:9790328) gene. Transgenic Arabidopsis thaliana plants (cv. Columbia) were grown from seeds provided by the Salk Institute. Transgenic rice (Oryza sativa cv. Dongjin) plantlets harboring the nptII (neomycin phosphotransferase II) gene conferring kanamycin resistance were also grown from seeds of nptII transgenic plants. Transgenic Spirodela polyrhiza plants were produced by transformation with the nptII gene. Transgenic Chlamydomonas reinhardtii lines were produced by transformation with the aminoglycoside phosphotransferase (aph7) gene for hygromycin selection. Porphyra tenera (red alga) samples were prepared from thallus grown in vitro. In vitro-cultured plant tissues or plants grown in a growth chamber were used for DNA extraction.
DNA extraction procedure
The DNA extraction procedure is summarized in Fig. 1a. In a 1.5-ml microfuge tube (Eppendorf, Hamburg, Germany), shoot or leaf tissue (10–15 mg; one or two leaf discs with a diameter of 1 cm) is homogenized with a pestle for 1 min in 150–300 μl of 5% Chelex 100 (Bio-Rad, USA). The tissue is vortexed for 10 s, incubated in boiling water for 5 min, then vortexed again for 10 s, and finally centrifuged at 13,000 rpm for 1 min. The supernatant can then be used as template for PCR amplification.

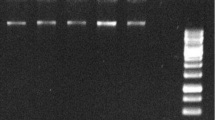
Rapid extraction of DNA from tobacco plant and PCR amplification of transgene. a An novel rapid procedure for DNA isolation using Chelex 100. b Gel electrophoresis of DNA extracted from tobacco leaf with the newly developed Chelex 100 method. c PCR amplification of TBP19 from transgenic tobacco plants using the method shown in a. LN2+grinding+boiling PCR amplification using the method with LN2 (liquid nitrogen) treatment, Grinding+boiling PCR amplification using the grinding and boiling method, Boiling PCR amplification using grinding and boiling method without homogenizing step, Grinding PCR amplification using the grinding and boiling method without incubation at 100°C. M 1-kb DNA size marker, WT wild-type tobacco, lanes 1–3 transgenic plants, P positive control, isolated DNA using the Qiagen kit for transgenic plants
PCR analysis
In our experiments, an aliquot of extracted DNA (0.5–2 μl) was used as template for the PCR. DNA, primers, Taq polymerase, and reaction buffers were mixed for PCR cycling (GeneAmp PCR system 9700; Applied Biosystems, Foster City, CA) and amplified under the following cycling conditions: one cycle at 94°C for 5 min, followed by 30–40 cycles of 94°C for 30 s, 52–58°C for 30 s, and 72°C for 1–2 min, and terminated by a final 5-min extension at 72°C. The following primer pairs were used to amplify the target genes: TBP19-F (5′-CGCCATGGAACGAGCTATACA-3′) and TBP19-R (5′-CCTCTAGAAGGTCTCAGTACCT-3′) amplified a 0.5-kb TBP19 fragment; NPTIIF (5′-GAGGCTATTCGGCTATGACTG-3′) and NPTIIR (5′-ATCGGGAGCGGCGATACCGTA-3′) amplified a 0.7-kb nptII fragment; btbP-5 (5′-CCACTCGAGCTTGTGATCGCACT-3′) and Aph7R2 (5′-TTCCGGTCGGTCGTGCCGTCCAT-3′) amplified a 0.6-kb aph7 fragment; PtHSP70R3 (5′-ACAGGAGCCGACGCGTGCCA-3′) and PtHSP70P1KF (5′-TTGGATCCGAACCTGCCCCCGGGT-3′) amplified a 1.1-kb heat shock protein (HSP 70) fragment from P. tenera; PtBtub (31 5′-CTCCGASACSAGGTCRTTCAT-3′) and PtBtub51 (5′-AACAACTGGGCYAAGGGSCA-3′) amplified a 1-kb beta tubulin (bTub) fragment from P. tenera.
Results and discussion
Optimization of rapid DNA extraction for PCR
To develop a rapid protocol for plant DNA extraction for PCR, we modified a colony PCR protocol used for Chlamydomonas (Cao et al. 2009) by adding a homogenizing step and removing a cooling step (Fig. 1a). The modified protocol successfully extracted DNA from freshly harvested and liquid nitrogen-frozen tobacco leaf discs, demonstrating that DNA obtained from frozen plant samples using this method is suitable for PCR amplification. Homogenization of plant tissue is considered to be critical for successful PCR amplification after rapid DNA extraction. To test this assumption, we evaluated PCR efficiency after DNA extraction without the homogenization step and found that although the PCR did not always amplify transgenes without the homogenization step, the amplification itself appeared to be successful if the boiling time was extended or the number of PCR cycles was increased. Furthermore, at least a 5-min incubation in boiling water was required to produce rapid and consistent PCR amplification. We found that the step eliminated from the Chlamydomonas colony PCR protocol (chilling on ice) was not essential for PCR amplification of plant DNA: the PCR successfully amplified transgenes in all samples of DNA extracted without this step. Figure 1b, c shows the results of these experiments, illustrating the efficiency of the various modifications for extracting of tobacco plant DNA for PCR analyses.
PCR amplification of transgenes from various species of transgenic plants
Berthold et al. (1993) reported successful PCR amplification with Chlamydomonas and Arabidopsis DNA extracted using Chelex 100, ethanol, and autoclaving steps, but they failed to amplify a transgene in tomato. To develop a universal plant DNA extraction protocol, we tested the applicability of our modified Chelex 100 method with DNA from various species. In this study, we used our method to extract DNA from tomato tissues cultured in vitro and grown in a growth chamber; the DNA was subsequently tested by PCR amplification. We found that the transgene from the DNA of transgenic tomato plants was successfully amplified (Fig. 2a; lanes 12, 14, 16, 61, and 64) and that the samples without a band were from nontransgenic plants. This result was confirmed by PCR amplification of DNA isolated with a Qiagen kit (DNeasy Plant Mini kit, Qiagen, Valencia, CA) (Fig. 2a; lanes 11 and 63). These findings indicate that our rapid DNA extraction method produces high-quality DNA from tomato plant samples for PCR.

PCR amplification of transgenes from tomato, potato, Arabidopsis thaliana, rice, strawberry, Spirodela polyrhiza, Chlamydomonas reinhardtii, and Porphyra tenera. Plant DNA was extracted using the method shown in Fig. 1a. a PCR screening of tomato plants transformed with the TBP19 gene (0.5 kb). b PCR screening of potato plants transformed with the TBP19 gene (0.5 kb). c PCR amplification of the nptII gene (0.7 kb) from T-DNA mutant lines of A. thaliana (lanes: 1 CHI-OX-4, 2 CHS-OX21, 3 F3′H-OX-24, 4 LDOX-OX-15, 5 mylo56-4BC-7, 6 Salk-112104c). d PCR amplification of the nptII gene (0.7 kb) from transgenic rice. e PCR amplification of the nptII gene (0.7 kb) from transformed strawberry plants. f PCR amplification of the nptII gene (0.7 kb) from transformed S. polyrhiza. g PCR screening of Chlamydomonas lines transformed with the aph7 gene (0.6 kb). h PCR amplification of HSP70 (1.1 kb) and bTub (1 kb) genes from P. tenera (1C HSP70 amplified from DNA obtained by rapid extraction, 2C bTub amplified from DNA was obtained by rapid extraction, 1g HSP70 amplification from DNA by the Qiagen kit and cetyl trimethylammonium bromide extraction, 2g bTub amplification from DNA by Qiagen kit and CTAB extraction). Lanes 1–6 independent transgenic plants, WT wild type, P positive control, isolated DNA using Qiagen kit from transgenic plant
This method was evaluated using a diverse range of plant species including potato, Arabidopsis, rice, strawberry, S. polyrhiza (hydrophyte), C. reinhardtii (green algae), and Porphyra tenera (red alga), and the genes of interest were successfully amplified from all species tested (Fig. 2 ). Although the amplification of some DNA samples produced only faint PCR bands following electrophoresis in an agarose gel, increasing the number of PCR cycles or the quantity of the DNA template produced stronger bands, whereas the same conditions still produced no signal from the DNA of wild-type plants lacking the transgene.
Conventional methods used to extract DNA from plant and algal species for PCR, such as the cetyl-trimethylammonium bromide procedure, require many tedious steps to remove the polysaccharides that act as potent PCR inhibitors (Furukawa and Bahavanandan 1983). However, PCR amplification of DNA extracted using our method successfully amplified nptII from a transgenic strawberry plant and HSP70 and bTub from P. tenera, indicating that 5% Chelex 100 removed or sufficiently reduced the activity of PCR inhibitors.
Our method was successfully applied to higher plants, hydrophytes, and algae. This method differs from existing protocols for plant DNA extraction in that we were able to omit the phenol/chloroform extraction and ethanol precipitation steps. The supernatant containing the DNA template for PCR amplification, which presumably contains carbohydrates and denatured protein, had no apparent inhibiting effect on the PCR reaction. A 0.5- to 2-μl aliquot out of the total 150–300 μl supernatant was sufficient for PCR amplification. This method requires little manipulation and can easily be applied to hundreds of samples. The entire extraction procedure requires <15 min to prepare the DNA for PCR, and culture or growing time can be saved because only a small tissue sample is needed. In addition, the supernatant contains degraded RNA (Fig. 1a), suggesting the possibility that the protocol can be modified to improve RNA quality and then used for reverse transcription (RT)-PCR.
In conclusion, we have developed a rapid and simple method for DNA extraction using the chelating resin Chelex 100. Our technique prepares DNA from various higher plants and algal species that is suitable for use in PCR analyses. It differs from the conventional DNA extraction method using Chelex 100 by omitting a number of time-consuming and tedious steps normally required for DNA extraction from plant material. As shown here, the method produces reliable results and can be used routinely in the laboratory for a variety of plant and algal species.
References
Berthold DA, Best BA, Malkin R (1993) A rapid DNA preparation for PCR from Chlamydomonas reinhardtii and Arabidopsis thaliana. Plant Mol Biol Rep 11:338–344
Cao M, Fu Y, Guo Y, Pan J (2009) Chlamydomonas (Chlorophyceae) colony PCR. Protoplasma 235:107–110
Furukawa K, Bahavanandan VP (1983) Influences of anionic polysaccharides on DNA synthesis in isolated nuclei by DNA polymerase alpha: correlation of observed effects with properties of polysaccharides. Biochim Biophys Acta 740:466–475
Holmes AR, Cannon MG, Shepherd MG (1994) Detection of Candida albicans and other yeast in blood by PCR. J Clin Microbiol 32:228–231
Panaccio M (1991) PCR based diagnosis in the presence of 8% (v/v) blood. Nucleic Acid Res 19:291–292
Tebbe CC, Vahjen W (1993) Interference of humic acids and DNA extracted from soil in detection and transformation of recombinant DNA from bacteria and yeast. Appl Environ Microbiol 59:2657–2665
Tsai YL, Olson BH (1992) Rapid method for separation of bacterial DNA from humic substances in sediments for polymerase chain reaction. Appl Environ Microbiol 58:2292–2295
Walsh PS, Metzger DA, Higuchi R (1991) Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Ward AC (1992) Rapid analysis of yeast transformants using colony-PCR. Biotechniques 13:350
Acknowledgments
This work was supported by a grant (Code 20070301034020) to WJJ from the BioGreen 21 Program, the Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
HwangBo, K., Son, S.H., Lee, J.S. et al. Rapid and simple method for DNA extraction from plant and algal species suitable for PCR amplification using a chelating resin Chelex 100. Plant Biotechnol Rep 4, 49–52 (2010). https://doi.org/10.1007/s11816-009-0117-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-009-0117-4