
Abstract
Single-strand conformation polymorphism (SSCP) is a reproducible and sensitive method for the detection of genetic polymorphisms and mutations in a wide range of polymerase chain reaction (PCR) products. However, the applications of this technique are largely confined to a set of cumbersome optimizations. Herein, a non-time-consuming method for PCR-SSCP that can be conducted with minimum efforts and less technical expertise is presented. The main concept of this simplified technique was based on many optimizations that were conducted to ensure the highest possible sensitivity to detect genetic polymorphism without incorporating sophisticated equipment and tedious efforts. The optimum gel concentration, temperature, and time requirements were strictly adjusted so as not to be further modified before applying this method for genotyping purposes. Furthermore, minimized silver staining steps were combined with this method to further minimize time and effort. It was confirmed that the performed adjustments were not reduced the overall sensitivity of the technique. Therefore, the suggested method can be utilized to genotype a wide range of PCR products (mainly from 200 to 600 bp) without the need for further optimizations and modifications. This study proposes a rapid SSCP protocol for genotyping PCR products using simple, low-cost, and friendly to perform recipes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polymerase chain reaction–single-strand conformation polymorphism (PCR-SSCP) is a versatile method used in the genotyping of PCR products in a vast range of organisms. It allows the detection of the possible causative unknown single nucleotide polymorphism (SNP) and mutations that could not be identified by other commonly used methods, such as PCR-restriction fragment length polymorphism (RFLP) (Hashim and Al-Shuhaib 2019). PCR-SSCP has initially employed to identify the possible genetic variations within PCR amplicons (Orita et al. 1989). The main concept of PCR-SSCP is based on the melting of the double-stranded DNA by denaturation into single-stranded forms. In the separated state, the single-stranded DNA molecules tend to fold into particular three-dimensional (3D) conformations based on nucleic acid sequences. According to the sequence variations, the separated strands of DNA may accommodate different 3D structures and altered electrophoretic mobility in the vertical polyacrylamide gels. The reason for such altered mobility is the presence of an SNP that possibly alters the physical conformations of the denatured single strands in the polyacrylamide gel. This alteration often leads to a slightly tilted positioning of the mutant single-stranded molecules than these occupied by the normal molecules in the same gel (Gasser et al. 2006). Despite the development of high-throughput next-generation sequencing reactions (Rennert et al. 2016), their large-scale applications are confronted by their high cost. Alternatively, PCR-SSCP has been extensively used in genotyping of PCR amplicons in a wide range of organisms (Konstantinos et al. 2008; Mustafa et al. 2018; Bai et al. 2019; Albakri et al. 2020; Aljubouri et al. 2020b; Musafer et al. 2021). The low-cost PCR-SSCP method becomes more common due to many accumulated data that reported the ability of this technique to detect enormous nuclear and extra-nuclear genetic variations (Bandyopadhyay et al. 2010; Csikos et al. 2016). Although it can be applied to many genes for many organisms, PCR-SSCP cannot be used without conducting several technical modifications that may narrow its practical validity in genotyping reactions. The main difficulty in genotyping PCR products with SSCP is correlated with the exact conformation of a DNA fragment which cannot be determined under the effects of variable parameters (Tabit 2016). Thus, many optimization steps are usually employed to control various parameters interacting with the sensitivity of this technique, such as amplicons sizes, the porosity of the gels, loaded amplicons, power applied as well as other variable conditions. Many optimizations can be employed for PCR-SSCP through changing the gel concentration (8–14 %), the temperature of electrophoresis (4–20 °C), and the power applied (5–10 V/cm). Without being properly optimized, these variable conditions may reduce the overall sensitivity of genotyping experiments with this method. Furthermore, various pre-electrophoresis steps should also be conducted before loading PCR amplicons onto polyacrylamide gels. However, conducting such optimizations may add another layer of complexity to PCR-SSCP. Though PCR-SSCP allows sensitive discrimination among different conformers within an amplicon, its efficacy is largely hampered by the necessity to conduct tedious optimizations and the lack of experience (Cremonesi et al. 2012). Hence, it is mandatory to reduce the difficulty in performing PCR-SSCP taking into account not to compromise its overall sensitivity. Likewise, it is preferable to conduct PCR-SSCP without involving any previous optimizations and modifications. To do so, variable technical issues should be solved by providing an applicable recipe suitable for genotyping of the majority of PCR products. This study is therefore aimed to present an efficient and simplified PCR-SSCP protocol that can be conducted directly to genotype a wide range of PCR products using ready-to-use recipes without wasting time and effort.
Materials and methods
Biological samples
This work was approved by the scientific committee of Al-Qasim Green University, Al-Qasim, Iraq (approval no. 2334, dated 22-04-2016). The experimental procedures of animal experimentations for this study were approved by the Animal Committee of Al-Qasim Green University, Babil, Iraq. Through many efforts, a variety of mammals, birds, and microbiological organisms were genotyped by this protocol. The included species were humans (n = 300), sheep (n = 224), goats (n = 91), Japanese quails (n = 223), and African ostriches (n = 69). This method was also applied on Aspergillus flavus sequences (n = 122). All included human subjects were signed informed consent before donating their blood. The blood samples of sheep and goats were collected from Barakat Abu al Fadhl Al-Abbas Station for raising sheep (Al-Khafeel co., Karbala, Iraq). Feathers of African ostriches were collected from Ejaimy husbandry facilities, located in Mahawil and Musaib areas, in Babil and Baghdad governorate respectively. Samples of A. flavus were isolated from the stored rice staff in local markets of Kufa and Babil governorates according to the procedure described by (Al-Shuhaib et al. 2018b).
Genomic DNA extraction
Concerning genomic DNA (gDNA) from human subjects, sheep, and goats, a universal and rapid salting-out procedure for DNA extraction was applied (Al-Shuhaib 2017). Concerning gDNA extraction from mammals, about 2 mL of blood samples were collected from all investigated subjects and stored in anti-coagulation tubes and taken to the laboratory in an icebox and kept at 4 °C until use. All efforts were made to minimize any discomfort during sample collection. Concerning gDNA extraction from African ostriches, 1 cm at the bottom of the healthy feature was used for the extraction procedure following the instructions mentioned in (Al-Shuhaib et al. 2018a). Genomic DNA extraction was also manually extracted from the blood samples of Japanese quails according to the procedure mentioned in (Al-Shuhaib 2018). The gDNA of the analyzed A. flavus isolates were extracted following to details mentioned by (Cenis 1992). In all extraction procedures, the integrity of the extracted DNA was evaluated using agarose gel electrophoresis, while the purity and quantity of the extracted gDNA were assessed using a Nanodrop spectrophotometer.
PCR conditions
To validate the presented PCR-SSCP protocol in terms of the upper and lower limits for genotyping purposes, many primers with variable lengths were included in these validations. Using the NCBI primer blast server (Ye et al. 2012), PCR specific primers’ pairs were designed to incorporate variable loci within humans (Mohammed et al. 2021), sheep (Aljubouri et al. 2020a, b; Al-Thuwaini et al. 2020; Aljubouri and Al-Shuhaib 2020), goats (Al-Shuhaib et al. 2019), quails (Hussein et al. 2020), ostriches (Al-Shuhaib et al. 2018a), as well as the fungal microorganism A. flavus (Albakri et al. 2020). The details for these designed PCR primers’ pairs are described in Table 1. The PCR amplification reactions were performed using Bioneer PCR premix (Daejeon, South Korea). The standard PCR protocol was initiated by one cycle of denaturation at 94 °C for 4 min, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing for 45 s, and elongation at 74 °C for 45 s, and was concluded with a final extension at 72 °C for 5 min. The specificities of PCR products were verified by electrophoresis on 1.5 % agarose gel.
SSCP conditions
Several conditions were adjusted to get the best-optimized parameters that could be followed without losing time and effort. In this protocol, temperature, voltage, time, gel concentrations, electrophoresis module, and staining conditions were optimized altogether to get the highest possible resolution. After conducting many optimizations, the best rapid, affordable, and most sensitive steps of PCR-SSCP were obtained. In this protocol, each PCR amplicon was treated with an equal volume of SSCP denaturing-loading buffer (95 % formamide, 0.05 % xylene cyanol, 0.05 % bromophenol blue, and 20mM EDTA, pH 8). After denaturation for 7 min, 2 µL of each PCR product was immediately placed on ice and frozen for about 10 min. Only 2 µL of each denatured sample was loaded in 0.1 cm neutral polyacrylamide (37.5 acrylamide:1 bisacrylamide) gels containing 7 % glycerol in 1× TBE (tris-borate-EDTA) buffer. Vertical gel electrophoresis experiments were conducted in a mini-wide module (10 cm length, and 20 cm width) polyacrylamide gels to incorporate the largest number of samples per run. After conducted a series of PCR-SSCP genotyping experiments, the electrophoresis conditions of PCR amplicons were optimized to 8 % gel concentration while running time was adjusted to 3.5–4.0 h, 200 V, and 100 mA.
Silver staining of gels
Gels were fixed and stained as described by Byun et al. (2009) with several modifications to reduce time and effort. Briefly, the polyacrylamide gel was impregnated with a fixing-staining solution (10 % ethanol, 0.5 % glacial acetic acid, 0.2 % silver nitrate) for 10–20 min, and rinsed with distilled water. The desired SSCP bands were developed with a developer solution (3 % sodium hydroxide, 0.1 % formaldehyde) until sufficient resolution was obtained. The development of SSCP bands was not terminated and the silver-stained gels were placed immediately on a white lightbox and photographed.
DNA sequencing
Each detected genotype on the SSCP gel was subsequently exposed to sequencing reactions from both termini according to instructions of Macrogen laboratories (Macrogen, Geumchen, Seoul, Korea). The referring database of the nucleic acid sequences was retrieved from the national center for biotechnology information (NCBI) (https://www.ncbi.nlm.nih.gov). The sequenced SSCP genotypes were annotated by BioEdit ver, 7.1. (DNASTAR, Madison). Each detected variant was visualized using SnapGene Viewer ver. 4.0.4 (http://www.snapgene.com). Only clear electropherograms were considered in the observed variations in comparison with their retrieved corresponding reference sequences.
Results and discussions
The widespread utilization of PCR-SSCP has been restricted due to variable technical optimizations that reduced its feasibility of genotyping. This study has managed to solve the main technical issues that confined this technique via undergoing extended genotyping experiments on variable lengths of PCR amplicons. Accordingly, universal, simple, inexpensive, and ready to use recipes were formulated to perform PCR-SSCP genotyping without losing time, money, and effort. Using a high voltage protocol, distinct separations were observed among the amplified fragments of humans, sheep, goats, ostriches, quails, and fungal organisms. Two to three distinctive genotypes were resolved in the amplified PCR products among all investigated mammals, birds, and microorganisms (Fig. 1). Due to the potential of PCR-SSCP to detect the unknown mutations (Hashim and Al-Shuhaib 2019), it is mandatory to involve this protocol in clinical genetics alongside other broad agricultural and biological applications (Kostantinos et al. 2008). As in other tested organisms, our results indicated that the proposed protocol could also be applied for the genotyping of genetic diseases in the involved human participants. The results showed high sensitivity of the presented rapid PCR-SSCP protocol to identify the tiny differences among different genotypes in each particular status. The validity of each resolved genotype was confirmed by direct Sanger sequencing experiments, which were consistent with the observed heterogeneity among SSCP bands.

Validated efficiency of the presented rapid optimized PCR-SSCP in genotyping of mammals (human, sheep, and goats), birds (quails and ostriches), and the fungal (Aspergillus flavus) organisms
PCR-SSCP can detect nucleotide polymorphism between 200 and 600 bp fragments (Gasser et al. 2006). Therefore, variable lengths of PCR amplicons were used to validate the SNP detection ability of this protocol in a wide range of PCR products, starting from around 200 bp to more than 600 bp of lengths. Accordingly, lower genotyping potential can be expected for PCR fragments having less than 200 bp or more than 600 bp. In the first case (i.e. fragments with less than 200 bp), it is not usually feasible to utilize PCR-SSCP protocol to identify the unknown mutation in these short fragments due to the lower number of the expected variants. Meanwhile, it is worthwhile to design amplicons having more than 600 bp of lengths, i.e. COX1 with 689 bp and CYTB with 640 bp, to assess the utmost ability of this protocol to genotype such challenging lengths of PCR products. Though longer stretches of DNA have not been suggested in literature due to many technical limitations (Menounos and Patrinos 2010), the results of this study showed that our rapid protocol can also be used in PCR products with more than 600 bp lengths. Another layer of confirmation for the practical feasibility of this protocol was originated from the ability of the present PCR-SSCP protocol to perform genotyping of nuclear and mitochondrial sequences with parallel efficiency. This high power of separation provided an obvious role for the currently implemented tool in providing sufficient resolution among PCR amplicons without the need for any previous optimization for a vast range of sequences.
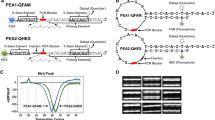
In this protocol, several conditions were adjusted, such as running time and temperature, power applied, gel concentration and thickness, and dimensions of electrophoresis format. These optimized conditions were combined all together in a final recipe that can easily be followed to genotype PCR products without the use of further unnecessary adjustments (Fig. 2).

The main modifications were made to perform sensitive genotyping with PCR-SSCP without prior optimizations. a preferred polyacrylamide gel components; 37.5 g bisacrylamide per 1 g bisacrylamide. b gel concentration, thickness, and module; the preferred conditions are 8 % of polyacrylamide molded in 10 cm length x 20 cm width gel dimensions in 0.1 cm gel thickness. c the running time, power applied, and temperature; the preferred electrophoresis running time is 3.5–4 h, and the preferred power applied is 200 V / 100 mA. Electrophoresis conditions were conducted at room temperature. d silver staining method; few steps of staining with silver nitrate are suggested to minimize time, money, and efforts without losing the efficiency of discrimination between genotypes
It is well known that PCR-SSCP requires an extended time of electrophoresis (Hayashi and Yandell 1993). But in the currently suggested protocol, no more than 4 h of continuous electrophoresis is required. It is usually acknowledged that the time of running gel should not exceed 23 °C. If the running temperature is between 23 and 27 °C, the running time should be reduced to 3.5 h. If the temperature of the laboratory is more than 27 °C, the running time should not be extended beyond 3 h. However, it is not desired to run this protocol at such higher temperature degrees due to the inability of some amplicons to withstand such extreme conditions without losing resolving power. In contrast to other PCR-SSCP protocols that could not be conducted without recirculating chillers (Aali et al. 2017), room temperature is the only temperature required to perform this protocol. However, the current method is optimally applicable at 20 °C. At a temperature around 30 °C, it is not recommended to extend the running time over 3 h as this could lead to the potential flowing of these bands out of the specified gel. Furthermore, the higher or lower temperatures could lead to a potential respective distortion or halted migrations of the DNA single-strand DNA bands. In both cases, higher ratios of the reduced resolution would be identified. Thus, higher or lower temperatures could interfere with the migration of the loaded samples and exhibit diverse effects on the speed of discrimination among PCR products. However, the recommended power (V/mA) in this rapid protocol is 10 V/cm, which is equal to 200 V / 100 mA in the mini-wide modules that are usually used in combination with the recommended running time in all cases (3 h, 3.5 h, and 4 h). By applying such high voltage at room temperature (An et al. 2011), the investigated PCR amplicons will be forced to be resolved efficiently without wasting more time to get the same genotyping outcomes. After conducting many genotyping experiments using variable gel concentrations ranging from 6 to 14 %, it was made sure that gel concentrations with more than 8 % would give unwanted proportional retardation of bands migration and adds more unnecessary extended time to resolve SSCP bands that can easily be resolved with the same efficiency using only 8 % gel concentrations (Petrov et al. 2013). Whereas the utilization of less than 8 % of gel concentration, the resolving power kept in the recommended 8 % gels would be reduced. The currently suggested protocol can be used with all types of mini-gel formats, which are usually available in the routine laboratories with 10 × 10 cm (length x width). However, we recommend running samples in wider gel formats with dual 10 × 20 cm (length x width) dimensions to accommodate a large number of investigated samples at a time. So that, this presented protocol provides further saving for times, chemicals, and efforts since the preparation of larger gel formats with 20 × 20 cm (length x width) are expensive and require tedious technical adjustments. In addition to the recommended dimensions of gel length and width, the common 0.1 cm of the gel thickness is also advocated. This thickness is usually available in routine labs, which however can easily be handled without losing the desired resolution. Using this protocol, other less-common gel concentrations (1.5 mm, 0.75 mm, or 0.5 mm) are not recommended or not suitable with our suggested recipes. Using the combination of these preferred running time and temperature, power applied, gel concentration and thickness, and dimensions of electrophoresis format, it could be possible to genotype PCR products to get the best results without losing sensitivity observed in the technically demanding and time-consuming PCR-SSCP protocols.
Conclusions
After employing many optimizations for PCR-SSCP genotyping experiments it was made sure that the presented recipe could be applied by any technician who has a minimum experience in post-PCR genotyping issues. Several parameters were optimized to get the highest resolution in terms of gel concentration, thickness, gel-format dimensions, and electrophoresis conditions. Furthermore, it was confirmed that this protocol is suitable for the majority of PCR products that are extended in length within the lower and upper resolution limits of SSCP. The currently described protocol was validated by many specific PCR primers’ pairs that were designed to amplify variable regions within an ample range of organisms. This is the first PCR-SSCP protocol that can be conducted for variable PCR products without the need for any optimization.
References
Aali M, Moradi-Shahrbabak H, Moradi-Shahrbabak M, Sadeghi M, Yousefi AR (2017) Association of the calpastatin genotypes, haplotypes, and SNPs with meat quality and fatty acid composition in two Iranian fat-and thin-tailed sheep breeds. Small Ruminant Res 149:40–51. https://doi.org/10.1016/j.smallrumres.2016.12.026
Albakri AH, Al-Shuhaib MBS, Alwan SL, AbdulAzeez S, Borgio JF (2020) Deleterious missense variants in the aflatoxin biosynthesis genes explain the low toxicity of Aspergillus flavus from infected rice. Microb Pathog 104605. https://doi.org/10.1016/j.micpath.2020.104605
Aljubouri TRS, Al-Shuhaib MBS (2020) Genotyping of mitochondrial D-loop sequences in three breeds of sheep. Biologia 76(1):203–211. https://doi.org/10.2478/s11756-020-00543-6
Aljubouri TRS, Al-Shuhaib MBS, Javadmanesh A (2020a) HMGA2 gene polymorphisms and their effects on main growth traits indices in Awassi and Karakul sheep. Agr Natr Resour 54:587–594. https://doi.org/10.34044/j.anres.2020.54.6.03
Aljubouri TRS, Hassan AF, Al-Shuhaib MBS, Mahyari SA (2020) Association of GnRH1 gene with growth traits in two breeds of sheep. Agric Nat Resour 54:587–594. https://doi.org/10.1007/s40003-020-00501-3
Al-Shuhaib MBSA (2017) A Universal, rapid, and inexpensive method for genomic DNA isolation from the whole blood of mammals and birds. J Genet 96(1):171–176. https://doi.org/10.1007/s12041-017-0750-6
Al-Shuhaib MBS (2018) A minimum requirements method to isolate large quantities of highly purified DNA from one drop of poultry blood. J Genet 97:e87–e94. https://doi.org/10.1007/s12041-018-0983-z
Al-Shuhaib MBS, Al-Kafajy FR, Badi MA, AbdulAzeez S, Marimuthu K, Al-Juhaishi HAI, Borgio JF (2018) Highly deleterious variations in COX1, CYTB, SCG5, FK2, PRL and PGF genes are the potential adaptation of the immigrated African ostrich population. Comput Biol Med 100:17–26. https://doi.org/10.1016/j.compbiomed.2018a.06.019
Al-Shuhaib MBS, Albakri AH, Alwan SH, Almandil NB, AbdulAzeez S, Borgio JF (2018) Optimal pcr primers for rapid and accurate detection of Aspergillus flavus isolates. Microb Pathog 116:351–355. https://doi.org/10.1016/j.micpath.2018.01.049
Al-Shuhaib MBS, Al-Thuwaini TM, Fadhil IA, Aljobouri TRS (2019) GHRL gene-based genotyping of ovine and caprine breeds reveals highly polymorphic intronic sequences in Awassi sheep with several RNA motifs. J Genet Eng Biotechnol 17(1):3. https://doi.org/10.1186/s43141-019-0004-5
Al-Thuwaini TM, Al‐Shuhaib MBS, Lepretre F, Dawud HH (2020) Two co‐inherited novel SNPs in the MC4R gene related to live body weight and hormonal assays in Awassi and Arabi sheep breeds of Iraq. Vet Med Sci :1–11. https://doi.org/10.1002/vms3.421
An XP, Song SG, Hou JX, Zhu CM, Peng JX, Liu XQ, Liu HY, Xiao WP, Zhao HP, Bai L (2011) Polymorphism identification in goat DGAT2 gene and association analysis with milk yield and fat percentage. Small Ruminant Res 100:107–112. https://doi.org/10.1016/j.smallrumres.2011.05.017
Bai L, Zhou H, Gong H, Tao J, Ma Q, Ding W, Hickford JGH (2019) Variation in the ovine KAP8-1 gene affects wool fibre uniformity in Chinese Tan sheep. Small Ruminant Res 178:18–21. https://doi.org/10.1016/j.smallrumres.2019.07.008
Bandyopadhyay S, Bera AK, Sikdar S, De S, Ghosh S, Rana T, Bandyopadhyay S, Dandapat P, Bhattacharya D (2010) Intra-species sequence variability in 28s rRNA gene of Oesophagostomum venulosum isolated from goats of West Bengal, India. Asian Pac J Trop Med 3:515–518. https://doi.org/10.1016/S1995-7645(10)60124-1
Byun SO, Fang Q, Zhou H, Hickford JGH (2009) An effective method for silver-staining DNA in large numbers of polyacrylamide gels. Anal Biochem 385:174–175. https://doi.org/10.1016/j.ab.2008.10.024
Cenis JL (1992) Rapid extraction of fungal DNA for PCR amplification. Nucleic Acids Res 20:2380. https://doi.org/10.1093/nar/20.9.2380
Cremonesi P, Pozzi F, Ricchi M, Castiglioni B, Luini M, Chessa S (2012) Identification of Prototheca species from bovine milk samples by PCR-single strand conformation polymorphism. J Dairy Sci 95:6963–6968. https://doi.org/10.3168/jds.2012-5785
Csikos A, Hodzic A, Pasic-Juhas E, Javor A, Hrković-Porobija A, Goletic T, Gulyas G, Czegledi L (2016) Applicability and sensitivity of PCR-SSCP method for milk species identification in cheese. Acta Aliment 45:69–76. https://doi.org/10.1556/066.2016.45.1.9
Gasser RB, Hu M, Chilton NB, Campbell BE, Jex AJ, Otranto D, Cafarchia C, Beveridge I, Zhu X (2006) Single-strand conformation polymorphism (SSCP) for the analysis of genetic variation. Nat Protoc 1:3121. https://doi.org/10.1038/nprot.2006.485
Hashim HO, Al-Shuhaib MBS (2019) Exploring the potential and limitations of PCR-RFLP and PCR-SSCP for SNP detection: A review. J Appl Biotechnol Rep 6(4):137–144. https://doi.org/10.29252/JABR.06.04.02
Hayashi K, Yandell DW (1993) How sensitive is PCR-SSCP? Human Mutat 2:338–346. https://doi.org/10.1002/humu.1380020503
Hussein T, Al-Shuhaib MBS, AL-Thuwaini TM (2020) Potential mitochondrial diversity role in the productivity of three lines of Japanese quails. Biodiversitas 21(5):2258–2265. https://doi.org/10.13057/biodiv/d210556
Konstantinos KV, Panagiotis P, Antonios VT, Agelos P, Argiris NV (2008) PCR–SSCP: A method for the molecular analysis of genetic diseases. Mol Biotechnol 38(2):155–163. https://doi.org/10.1007/s12033-007-9006-7
Menounos PG, Patrinos GP (2010) Mutation detection by single strand conformation polymorphism and heteroduplex analysis. In: Patrinos GP, Ansorge WJ (eds) Molecular Diagnostics, 2nd edn. Academic, Cambridge, pp 45–58
Mohammed AK, Al-Thuwaini TM, Al-Shuhaib MBS (2021) Single nucleotide polymorphism rs7908486 of the tcf7l2 gene is highly associated with obesity in the Iraqi population. Arch Biol Sci 73(1):39–45. https://doi.org/10.2298/ABS201213056M
Musafer KNJ, Huyop FZ, Ewadh MJ, Supriyanto E, Al-Thuwaini TM, Shuhaib MBS (2021) The single nucleotide polymorphisms rs11761556 and rs12706832 of the leptin gene are associated with type 2 diabetes mellitus in the Iraqi population. Arch Biol Sci 73(1):93–101. https://doi.org/10.2298/ABS210129005M
Mustafa KM, Ewadh MJ, Al-Shuhaib MBS, Hasan HG (2018) The in silico prediction of the chloroplast maturase k gene polymorphism in several barley varieties. Agriculture 64(1):3–16. https://doi.org/10.2478/agri-2018-0001
Orita M, Iwahana HKH, Hayashi K, Sekiya T (1989) Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci 86:2766–2770. https://doi.org/10.1073/pnas.86.8.2766
Petrov A, Tsa A, Puglisi JD (2013) Analysis of RNA by analytical polyacrylamide gel electrophoresis. Methods Enzymol 530:301–313. https://doi.org/10.1016/B978-0-12-420037-1.00016-6
Rennert H, Eng K, Zhang T, Tan A, Xiang J, Romanel A, Kim R, Tam W, Liu Y-C, Bhinder B (2016) Development and validation of a whole-exome sequencing test for simultaneous detection of point mutations, indels and copy-number alterations for precision cancer care. NPJ Genomic Med 1:1–11. https://doi.org/10.1038/npjgenmed.2016.19
Tabit FT (2016) Advantages and limitations of potential methods for the analysis of bacteria in milk: a review. J Food Sci Technol 53:42–49. https://doi.org/10.1007/s13197-015-1993-y
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL (2012) Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 18(13):134. https://doi.org/10.1186/1471-2105-13-134
Acknowledgements
This work was partially supported by Al-Qasim Green University. Authors would like to express their sincere thanks to all breeders of Barakat Abu al Fadhl Al-Abbas Station for raising sheep (Al-Khafeel co., Karbala, Iraq), and Ejaimy husbandry facilities (Mahawil and Musaib areas, in Babil and Baghdad, Iraq), for providing all necessary facilities during biological samples collection.
Author information
Authors and Affiliations
Contributions
MAB; conducted most of the experiments. MBSA; designed, supervised the work and wrote the manuscript. TRA; participated in the experiments and analyzed the data. TMA; co-supervised the work. HHD, THH, ATA, DA, MKAA, IAF, AHA; participated in the experiments. HOH and AMM; analyzed the data.
Corresponding author
Ethics declarations
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Badi, M.A., Al-Shuhaib, M.B.S., Aljubouri, T.R.S. et al. Rapid and optimized protocol for efficient PCR-SSCP genotyping for wide ranges of species. Biologia 76, 2413–2420 (2021). https://doi.org/10.1007/s11756-021-00776-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11756-021-00776-z