
Abstract
A simple, precise, accurate and selective method was developed and validated for determination of malondialdehyde (MDA) in olive oil. Separation was achieved on a reversed-phase C8 column using a mobile phase consisting of methanol/0.8 % phosphoric acid (10:90, v/v), at a flow rate of 1.0 ml/min and UV detection at 220 nm. This method was validated according to the requirements for new methods, which include accuracy, precision, selectivity, robustness, a limit of detection, limit of quantitation (LOQ), linearity and range. The current method demonstrated good linearity over the range of 0.5–1000 ppm of MDA with r 2 greater than 0.999. The recovery of MDA in olive oil ranged from 97.1 to 99.1 %. The method was selective where MDA was distinctly separated from other compounds of the oil with good resolution. The method was also precise where the RSD of the peak areas of replicate injections of MDA standard solution were less than 1 %. The degree of reproducibility of the results obtained as a result of small deliberate variations in the method parameters and by changing the analytical operators proved that the method is robust and rugged. The low LOQ of MDA (0.5 ppm) using this method enables quantitation of MDA at low concentration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipid oxidation is one of the major causes of oil spoilage. It leads to the development of various off-odors, generally called rancidity, and discoloration, which render oils unacceptable or reduce their shelf-life. In addition, oxidative reactions can decrease the nutritional quality of oils, and certain oxidation products are potentially toxic. Two types exist: primary and secondary oxidation products [1, 2]. Thus, the assessment of lipid peroxidation is usually performed by analyzing the secondary oxidation products, such as MDA. This distinctive compound has long been employed as a model compound for studying secondary degradation products of lipid peroxidation. MDA has been measured by thiobarbituric acid, which is called the TBARS method [3, 4]. The TBARS method involves the reaction of 2-thiobarbituric acid (TBA) with MDA in edible oils to produce a chromogen that can then be determined spectrophotometrically at 532–535 nm. This method's major problem is the lack of specificity, where TBA reacts with other products of lipid peroxidation such as hydroperoxides and conjugated aldehydes to generate substances that absorb at 535 nm, similar to the adduct of MDA and TBA. Thus, the analysis of MDA by the spectrophotometric method is subject to misinterpretation [5, 6]. Most of the MDA present in oils exists bound to other oil constituents, and very little of it exists in the free form. Thus, acid must be added to the oil to be analyzed in order to liberate the MDA.
There are many methods in the scientific literature for determination of MDA. An HPLC method has been developed for determining the total MDA in vegetable oils after conversion of the MDA released from its precursor to the dansyl-pyrazole derivative [7]. In another HPLC method [8], the quantitation of MDA in aqueous distillates from freeze-dried chicken meat was determined using a mixed mobile phase of 1 % acetic acid and acetonitrile (15/85; v/v) with a UV detector at a level of 10−6 mol l−1. In another HPLC method [9], the determination of MDA in vegetable oils was obtained using a mobile phase of 1 % acetic acid and acetonitrile (85/15; v/v). Karatas et al. (2002) determined free MDA in human serum by HPLC using a mobile phase consisting of 30 mM potassium dihydrogen phosphate–methanol (65/35, v/v) at a flow rate of 1.5 ml min−1 [10].
The scope of the present work was to develop a simple, accurate, precise and selective method for MDA determination in olive oil using HPLC with a UV detector. Additionally, the method needed to be sensitive with a low limit of detection (LOD) and limit of quantitation (LOQ), where low concentrations of MDA could be determined as its concentration in oil is low. The objective of this work was therefore to develop and validate a sensitive, selective, precise, accurate, robust, rugged and linear (with wide dynamic range) method for the determination of MDA in olive oil. HPLC with a UV detector and isocratic elution method were used in the current work for MDA determination in olive oil. The method is simple with the reversed-phase mode being used with isocratic elution and using a UV detector, which is available in most analytical laboratories. Validation of the method was conducted in accordance with the requirements of new methods, which include linearity and range, accuracy, precision, selectivity, robustness, a limit of detection and limit of quantitation.
Methods
Chemicals
Methanol of HPLC grade was from J.T. Baker (NJ, USA). The phosphoric acid and malondialdehyde standard (HPLC grade) were purchased from Merck (Darmstadt, Germany). The olive oil samples were obtained from a local Palestinian market.
Apparatus
An HPLC system (Merck Hitachi LachromeElite HPLC system, Japan) with an L-2130 pump, L-2200 autosampler, L-2300 column oven and L-2490 UV detector was employed. Ezochrom Elite software was used. The C8 and C18 columns (5 µm, 150 mm length × 4.6 mm I.D.) were from Waters Corp. (Milford, MA, USA).
HPLC Conditions
UV detection was employed at 220 nm, isocratic elution was used at a flow rate of 1.0 ml/min, and the injection volume was set to 20 µl.
Preparation of the Standard and Sample Solutions
Solutions for Linearity and Range
Stock standard solution of MDA with a concentration of 1000 ppm was prepared by dissolving 100 mg of MDA in 100 ml of methanol. Six solutions of MDA with concentrations of 0.5, 5, 100, 300, 500 and 800 ppm were prepared from the stock standard solution by dilution using the mobile phase as diluent.
Solutions for Recovery of MdA
For determination of the recovery of MDA from olive oil, three solutions of MDA spiked in olive oil at three concentrations (5.0, 100.0 and 1000.0 ppm) were prepared. Then, MDA was extracted from the spiked oil samples as follows: 10 ml of trichloroacetic acid solution (7.5 % prepared in 0.1 M HCl solution) was added to 5 g of oil sample, homogenized for 2 min and centrifuged at 2000 rpm for 5 min. Then the aqueous layer was taken and used for analysis by HPLC. The solutions used for the recovery study were also used for the precision study.
Solutions for the LOD and LOQ
To determine the LOD and LOQ of MDA using this method, solutions with low concentrations (0.1, 0.2, 0.5 and 1.0 ppm), which were expected to produce a response of 3–20 times baseline noise, were prepared. The LOD was selected as the concentration of MDA that gave a signal-to-noise ratio of 3–10, while the LOQ was selected as the concentration that gave a ratio of 10–20.
Results and Discussion
Method Development
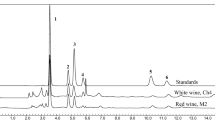
Preliminary studies involving trying C8 and C18 reversed-phase columns and testing several mobile phase compositions were conducted for the separation of MDA from other compounds present in olive oil with good chromatographic parameters (e.g., minimized peak tailing, good symmetry and good resolution between MDA and adjacent peaks). A C8 column (5 µm, 150 × 4.6 mm I.D.) as a stationary phase with a mobile phase of methanol/0.8 % phosphoric acid (10:90 v/v) at a flow rate of 1.0 ml/min and a detection wavelength of 220 nm afforded the best separation of MDA. Phosphoric acid in the mobile phase gave sharp peaks for MDA, while the mobile phase without phosphoric acid gave very broad peaks (low theoretical plates) with very poor resolution. Figure1a shows a chromatogram of a standard solution of MDA with a retention time of about 5.3 min, while Fig. 1b shows a chromatogram of MDA in a sample of olive oil that was force-degraded by heating the oil in oven at 70 °C for 2 days to enhance the oxidation of the oil.

Chromatogram of MDA analyzed by the current method: (a) Standard solution of MDA. (b) Sample of olive oil placed in an oven at 70 °C for 2 days. Mobile phase: methanol/phosphoric acid 0.8 % (10:90 v/v), flow rate 1.0 ml/min, injection volume 20 µl. Column: C8 (5 µm, 150 mm length × 4.6 mm I.D.), UV detection: 220 nm
Method Validation
After method development, validation of the method for MDA was performed in accordance with the requirements for new methods, which include accuracy, precision, selectivity, robustness, linearity and range, the LOD and LOQ.
Linearity and Range
To evaluate the linearity of the current method for the determination of MDA, different calibration standards of MDA were analyzed by HPLC-UV, and the responses were recorded. A plot of the peak areas versus concentration (in ppm) of MDA was found to be linear in the range of 0.5–1000 ppm with r 2 >0.999. This result demonstrated the linearity of this method over a wide dynamic range.
Accuracy (Percentage Recovery)
For determination of the percentage recovery of MDA in olive oil, it was spiked in olive oil at three concentration levels (5.0, 100.0 and 1000.0 ppm), followed by extraction of MDA from the oil and analysis by HPLC-UV. The average recovery for each level was calculated by the proportion of the area of the peak of MDA resulting from the spiked solution to the area of the peak resulting from a standard solution. The average recovery and RSD for each level were calculated. The results showed that the current method has good recovery (from 97.1 to 99.1 %) for MDA at the three concentration levels studied (5.0, 100.0 and 1000.0 ppm), with a relative standard deviation (RSD) lower than 1.0 %; see Table 1.
Precision
Repeatability
Repeatability of the current method for determination of MDA was evaluated by calculating the RSD of the peak areas of six replicate injections of the three standard solutions at three concentrations (5.0, 100.0 and 1000.0 ppm), and it (RSD) was found to be less than 1.0 % (Table 1). These results show that the current method for MDA determination is repeatable.
Intermediate Precision (Ruggedness)
Intermediate precision of the current method was evaluated by calculating the % recovery of MDA at three concentration levels (5.0, 100.0 and 1000.0 ppm) by another analyst on a different day. Results of this study showed that the % recovery obtained by the second analyst was comparable to that obtained by the main analyst and ranged from 98.2 to 99.4 % (Table 1), indicating that this method is rugged.
Selectivity
Selectivity of the current method was demonstrated by the good separation of MDA from other compounds present in olive oil with good resolution (resolution between the MDA peak and adjacent peak was 3.8, as shown in Fig. 1b, which shows a chromatogram of MDA in an oxidized olive oil sample that was prepared by storing an olive oil sample at 70 °C for 2 days).
Robustness
Robustness of the current method was investigated by analysis of MDA using the same developed method as in this study but deliberately changing one chromatographic condition each time. The chromatographic conditions that were changed were (1) flow rate (0.8 and 1.2 ml/min vs. the original flow rate of 1.0 ml/min), (2) the volume fraction of methanol (8 and 12 % vs. the original percentage of 10 %) and (3) wavelength (218 and 222 nm vs. the original wavelength of 220 nm). Results showed that separation was not affected by slightly changing the chromatographic conditions where resolution between the MDA and adjacent peak remained at about 3.8. Additionally, the recovery of MDA at three concentrations was not significantly affected by changing the chromatographic conditions (flow rate, % of methanol and wavelength); see Table 2.
Limit of Detection (LOD) and Limit of Quantitation (LOQ)
The LOD and LOQ of MDA using this method were determined by preparing dilute solutions of MDA (0.1, 0.2, 0.5 and 1.0 ppm), injecting them into the liquid chromatograph and recording the signal-to-noise ratio (S/N) for the MDA peak at each concentration. The LOD was selected to be the concentration that gave an S/N ratio between 3 and 10, while the LOQ was selected to be the concentration that gave an S/N ratio between 10 and 20. Results showed that the LOD and LOQ of MDA are 0.2 and 0.5 ppm, respectively. The low LOD and LOQ permit the determination of MDA in olive leaves at low concentration.
Conclusion
A simple, accurate, precise and selective HPLC method was developed and validated for the determination of MDA in olive oil. The method is linear for the determination of MDA with a wide dynamic range (0.5–1000 ppm). This method is also accurate where the % recovery of MDA is within 97.1–99.1 %. Precision of the method is confirmed by a low RSD of replicate injections of MDA. The method showed good separation of MDA from other compounds in olive oil with good resolution. The low LOD and LOQ of MDA enable the detection and quantitation of MDA in olive oil at low concentrations.
References
Griffiths G, Leverentz M, Silkowski H, Gill N, Sanchez-Serrano JJ (2000) Lipid hydroperoxide levels in plant tissues. J Exp Bot 51:1363–1370
Jasna L, Milan M, Marija M, Marijan K, Marta K, Vlasta B (2008) Measurement of malondialdehyde (MDA) level in rat plasma after simvastatin treatment using two different analytical methods. Periodicum Biologorum 110:63–67
Tsaknis J, Lalas S, Evmorfopoulos E (1999) Determination of malondialdehyde in traditional fish products by HPLC. Analyst 124:843–845
Mostaghim T, Ezzatpanah H, Boojar MM, Givianrad MH, Hosseini E, Azizinejad R (2013) The effect of various deep frying methods on malondialdehyde content of potato chips. Ann Biol Res 4:46–52
Draper HH, Hadley M (1990) Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol 186:421–431
Kishida E, Oribe M, Mochizuki K, Kojo S (1990) Determination of malondialdehyde with chemical derivatization into the pyrimidine compound and HPLC. Biochim Biophys Acta 1045:187–188
Hirayama T, Yamada N, Nohara M, Fukui S (1983) High performance liquid chromatographic determination of malondialdehyde in vegetable oils. J Assoc Off Anal Chem 66:304–308
Kakuda Y, Stanley DW, van de Voort FR (1981) determination of TBA number by high performance liquid chromatography. J Am Oil Chem Soc 58:773–775
Tsaknis J, Lalas S, Hole M, Smith G, Tychopoulos V (1998) An HPLC rapid Method of determining malonodialdehyde (MDA) for evaluation of rancidity in edible oils. Analyst 123:325–327
Karatas F, Karatepe M, Baysarb A (2002) Determination of free malondialdehyde in human serum by high-performance liquid chromatography. Anal Biochem 311:76–79
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Al-Rimawi, F. Development and Validation of a Simple Reversed-Phase HPLC-UV Method for Determination of Malondialdehyde in Olive Oil. J Am Oil Chem Soc 92, 933–937 (2015). https://doi.org/10.1007/s11746-015-2664-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-015-2664-x