
Abstract
Simulated gastrointestinal hydrolysis of hemp seed proteins using pepsin and pancreatin followed by membrane ultrafiltration fractionation yielded fractions with peptide sizes of <1, 1–3, 3–5, and 5–10 kDa. Analysis of in vitro antioxidant properties showed that the hemp seed protein hydrolysate (HPH) exhibited a significantly weaker (p < 0.05) scavenging of 2,2-diphenyl-1-picrylhydrazyl (DPPH) radicals when compared to the fractionated peptides. Metal chelation activity of the HPH was significantly greater (p < 0.05) than the activities of fractionated peptides. Fractionation of the HPH led to significant (p < 0.05) improvements in ferric reducing power, DPPH, and hydroxyl radical scavenging radical activities but decreased metal chelation capacity. Peptide fractions with longer chain lengths (3–5 and 5–10 kDa) had better metal chelation and ferric reducing power than the <1, and 1–3 kDa fractions. HPH and all the peptide fractions significantly inhibited (p < 0.05) linoleic acid oxidation when compared to the control. Glutathione (GSH) had significantly greater (p < 0.05) ferric reducing power, and scavenging of hydroxyl and DPPH radicals when compared to HPH and fractionated peptides. In contrast, HPH and peptide fractions >3 kDa had significantly higher (p < 0.05) metal chelation activity than GSH. The results show the potential use of HPH and peptide fractions of defined size for the treatment of oxidative stress-related diseases.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Industrial hemp (Cannabis sativa L.), widely cultivated in China and Canada, is a very important plant for food, fiber, and medicine. Hemp seed, a by-product obtained during the commercial utilization of the plant fiber contains over 30% oil and 25% of high quality protein [1]. Industrial hemp seed, which contains a low level (~0.3%) of δ-9-tetrahydrocannabinol (THC) is now legally grown in Canada. Hemp seed has been used in the development of numerous products for the cosmetics, therapeutic, functional food, and nutraceutical industries [2]. Hemp seed is also a rich source of polyunsaturated fatty acids, especially linoleic (ω-6) and α-linolenic (ω-3) acids, and proteins which contain all of the essential amino acids in nutritionally sufficient amounts for FAO/WHO suggested requirements for infants or children [3, 4]. The physicochemical and functional properties of hemp seed protein isolate (HPI) have been evaluated and compared with those of soy protein isolate (SPI): HPI had similar or higher levels of essential amino acids than SPI except for lysine. In addition, hemp seed protein hydrolysate (HPH) produced using different proteases have been reported to possess antioxidant properties [5] but the work did not provide information on relationships with amino acid content of peptides. Antioxidant peptides have continued to be a research interest due to the potential contributions to human health and especially for the prevention and treatment of chronic diseases.
Oxidative stress occurs as a result of an imbalance between the production of free radicals, reactive oxygen species (ROS) and the scavenging ability of endogenous antioxidants [6]. Excessive production of ROS may damage membranes, proteins, enzymes, and DNA resulting in the development of chronic disease conditions [7, 8]. Enzymatic food protein-derived peptides, in comparison to synthetic compounds are believed to be safer natural antioxidants that can be used as protective agents to help the human body reduce oxidative damage and associated diseases [6]. The antioxidant properties of these hydrolysates largely depend on enzyme specificity, degree of hydrolysis, and the nature of the peptides released including molecular weight, amino acid composition, and hydrophobicity [9–11]. The antioxidant properties of peptides include their ability to scavenge free radicals, inhibit linoleic acid autoxidation, act as chelating agents of metal ions, or as reducing agents [5]. The presence of certain amino acids like, histidine, tyrosine, methionine, lysine, tryptophan, and proline increases the antioxidant potency of most food-derived peptides [12]. To the best of our knowledge, there is lack of information on the relationships of peptide size and amino acid composition with antioxidant activities of hemp seed protein-derived hydrolysates. The present work contributes to the growing scientific knowledge on the relationships between amino acid content or peptide size and antioxidant properties of food protein hydrolysates.
Though a previous work [5] had used different enzymes, there is a need to determine the potential liberation of antioxidant peptides during oral consumption of hemp seed proteins. Therefore, the objective of this work was to produce a simulated gastrointestinal HPH, fractionate the hydrolysate into peptides of different molecular weights, and evaluate these samples for multifunctional properties using various antioxidant evaluation systems, in vitro. Glutathione (GSH) was used for comparison since it is a peptide (similar to the HPH components) and has physiological relevance as a cellular antioxidant molecule in human tissues.
Materials and Methods
Materials
Defatted hemp seed meal, referred to as hemp seed protein powder (HPP), was purchased from Manitoba Harvest Fresh Hemp Foods Ltd (Winnipeg, MB, Canada). Pepsin (porcine gastric mucosa, EC 3.4.23.1), Pancreatin (porcine pancreas), 2,2-diphenyl-1-picrylhydrazyl (DPPH), Triton X-100, hydrogen peroxide, ethylenediaminetetraacetic acid (EDTA), ferrous sulphate, potassium ferricyanide, trichloroacetic acid (TCA), ammonium thiocyanate, linoleic acid, ferrous chloride, 1,10-phenanthroline, 3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-4′,4″-disulfide acid sodium salt (ferrozine), and GSH were purchased from Sigma–Aldrich (St. Louis, MO, USA) while other analytical grade reagents and ultrafiltration membranes (1, 3, 5, and 10 kDa molecular weight cut-off) were obtained from Fisher Scientific (Oakville, ON, Canada).
Methods
Preparation of Hemp Seed Protein Isolates
HPI was produced from HPP according to the method described by Tang et al. [3] with slight modifications as follows. Briefly, HPP was dispersed in deionized water (1:20, w/v), and the dispersion was adjusted to pH 10.0 with 2 M NaOH to solubilize the proteins. The resultant dispersion was stirred at 37 °C for 2 h followed by centrifugation (7,000g at 4 °C) for 60 min. The pellet was discarded, and the supernatant filtered with cheesecloth and adjusted to pH 5.0 with 2 M HCl to precipitate the proteins. Thereafter, the mixture was centrifuged (7,000g at 4 °C) for 40 min, the resultant precipitate was re-dispersed in deionized water, adjusted to pH 7.0 with 2 M NaOH and freeze-dried to produce HPI powder. Protein content of HPI was determined using the modified Lowry method [13].
Production of Hemp Seed Protein Hydrolysate (HPH) and Membrane Fractions
Prior to enzymatic hydrolysis, the green-colored HPI was decolorized by mixing 1 g with 10 mL of acetone. The mixture was stirred in the fume hood for 3 h and decanted followed by a second and third consecutive extraction of the residue. The resulting HPI was air-dried overnight in the fume hood and used for further studies. The decolorized HPI was enzymatically hydrolyzed according to a previous method [14]. Briefly, 5% (w/v, protein basis) of decolorized HPI slurry was heated to 37 °C and adjusted to pH 2.0 using 1 M HCl. Protein hydrolysis was initiated by the addition of pepsin (4% w/v, protein basis) and the mixture stirred for 2 h. After peptic hydrolysis, the reaction mixture was adjusted to pH 7.5 with 2 M NaOH, pancreatin (4% w/v, protein basis) was added and the mixture was incubated at 37 °C for 4 h. The enzymatic reaction was terminated by adjusting the mixture to pH 4.0 with 2 M HCl followed by heating to 95 °C for 15 min to ensure a complete denaturation of residual enzymes. The mixture was centrifuged (7,000g at 4 °C) for 30 min and the resulting supernatant was sequentially passed through ultrafiltration membranes with molecular weight cut-off (MWCO) of 1, 3, 5, and 10 kDa in an Amicon stirred ultrafiltration cell. The permeate fraction from each MWCO membrane was collected, lyophilized, and stored at −20 °C until needed for further analysis. The protein contents of the freeze-dried HPH fractions were also determined using the modified Lowry method [13]. The above digestion and fractionation protocols were performed in triplicate and the freeze-dried products combined, analyzed for protein content [13] and used for antioxidant assays.
DPPH Radical Scavenging Assay
The scavenging activity of HPH and its fractions against the DPPH radical was determined using a previously described method [14] with slight modifications for a 96-well clear flat-bottom plate. Peptide samples were dissolved in 0.1 M sodium phosphate buffer, pH 7.0 containing 1% (w/v) Triton X-100. DPPH was dissolved in methanol to a final concentration of 100 μM. Peptide samples (100 μL) were mixed with 100 μL of the DPPH solution in the 96-well plate to a final assay concentration of 1 mg/mL and incubated at room temperature in the dark for 30 min. The absorbance values of the control (A c) and samples (A s) were measured at 517 nm. The control consisted of sodium phosphate buffer in place of the peptide sample while GSH was used as the positive control. The percent DPPH radical scavenging activity of the samples was determined using the following equation:
Chelation of Metal Ions
The metal chelating activity was measured using a modified method of Xie et al. [15]. Peptide sample solution or GSH (final assay concentration of 1 mg) was combined with 0.05 mL of 2 mM FeCl2 and 1.85 mL double distilled water in a reaction tube. Ferrozine solution (0.1 mL of 5 mM) was added and mixed thoroughly. The mixture was then allowed to stand at room temperature for 10 min from which an aliquot of 200 μL was removed and added to a clear bottom 96-well plate. A control was also conducted by replacing the sample with 1 mL of double distilled water. The absorbance values of (A c) and (A s) at 562 nm were measured using a spectrophotometer and the metal chelating activity of the sample was compared to that of GSH. The percentage chelating effect (%) was calculated using the following equation:
Ferric Reducing Power
The reducing power of peptide samples was measured according to a previously reported method [16] which was modified as follows. Peptide samples (250 μL) dissolved in 0.2 M sodium phosphate buffer at pH 6.6 or double distilled water (control) were mixed with 250 μL of buffer and 250 μL of 1% potassium ferricyanide solution. The final peptide concentration in the assay mixture was 1 mg/mL. The resulting mixture was heated at 50 °C and incubated for 20 min. After incubation, 250 μL of 10% of aqueous TCA was added. Thereafter, 250 μL of peptide/TCA mixture was combined with 50 μL of 0.1% ferric chloride and 200 μL of water and allowed to stand at room temperature for 10 min. The solution was centrifuged at 1,000g and 200 μL of the supernatant transferred to a clear bottom 96-well plate. The absorbance of the supernatant was measured at 700 nm.
Hydroxyl Radical Scavenging
The hydroxyl radical scavenging activity was determined based on a previously reported method [17]. Peptide samples and 3 mM of 1,10-phenanthroline were separately dissolved in 0.1 M sodium phosphate buffer (pH 7.4). FeSO4 (3 mM) and 0.01% hydrogen peroxide were both separately dissolved in distilled water. An aliquot (50 μL) of peptide samples (equivalent to a final assay concentration of 1 mg/mL) or buffer (control) was first added to a clear, flat bottom 96-well plate followed by 50 μL of 1,10-phenanthroline and then 50 μL of FeSO4. To initiate the Fenton reaction in the wells, 50 μL of hydrogen peroxide was added to the mixture, covered and incubated at 37 °C for 1 h with shaking. The absorbance was measured using a spectrophotometer at 536 nm at 10 min intervals for 1 h. The hydroxyl radical scavenging activity was calculated using the reaction rate (ΔA/min) equation below:
Inhibition of Linoleic Acid Oxidation
Linoleic acid oxidation was measured using the method described by Li et al. [8]. Peptide samples (final concentration of 1 mg/mL) were dissolved in 1.5 mL of 0.1 M sodium phosphate buffer (pH 7.0) and the mixture added to 1 mL of 50 mM linoleic acid dissolved in 99.5% ethanol. For the control assay, 1.5 mL of buffer was added to the ethanolic linoleic acid solution. The mixtures were kept at 60 °C in the dark for 7 days. At 24-h intervals, 100 μL of the assay solution was mixed with 4.7 mL of 75% aqueous ethanol, 0.1 mL of ammonia thiocyanate (30% w/v) and 0.1 mL of 0.02 M ferrous chloride dissolved in 1 M HCl. This solution (200 μL) was added to a clear bottom 96-well plate and the degree of color development was measured using the spectrophotometer at 500 nm after 3 min incubation at room temperature. An increased absorbance implied an increase in the level of linoleic acid oxidation.
Amino Acid Composition Analysis
The amino acid profiles of the samples were determined in duplicate (two separately digested and fractionated samples) using an HPLC system after samples were hydrolyzed with 6 M HCl [18]. The cysteine and methionine contents were determined after performing acid oxidation [19] and the tryptophan content was determined after alkaline hydrolysis [20].
Statistical Analysis
Antioxidant assays were conducted in triplicate and analyzed by one-way analysis of variance (ANOVA). The means were compared using Duncan’s multiple range test and significant differences accepted at p < 0.05.
Results and Discussion
Enzymatic Hydrolysis of Hemp Seed Protein Isolate and Ultrafiltration
In this study, HPI was decolorized with acetone to remove polyphenolic compounds that could interfere with accurate determination of antioxidant properties of the protein hydrolysates. This is because low molecular weight polyphenols have antioxidant properties, which could confound interpretation of the results. HPI was hydrolyzed consecutively with pepsin and pancreatin followed by sequential fractionation of the hydrolysate by ultrafiltration using 1, 3, 5, and 10 kDa MWCO membranes to produce peptide fractions with varying molecular weights. Pepsin and pancreatin enzymes were used in this study to mimic gastrointestinal protein digestion in human beings. Membrane fractionation resulted in enriched peptides with narrow molecular weight range (<1, 1–3, 3–5, and 5–10 kDa) and increased homogeneity of the peptides, when compared to the original HPH. In previous studies, membrane fractionation of protein hydrolysates resulted in fractions with better bioactive properties than their parent hydrolysates [14, 21]. The HPH had a protein content of 90% whereas the <1, 1–3, 3–5, and 5–10 kDa membrane ultrafiltration fractions contained 64, 85, 92, and 90% protein contents, respectively. The lower protein content of the <1 kDa permeate suggests that low molecular weight non-protein components such as salts and soluble sugars are present.
Amino Acid Composition
The amino acid compositions of HPI, HPH, and membrane fractions are shown in Table 1. The amino acid content of the HPI was very similar to previously reported data [3] and shows very high levels of arginine. The results indicated that amino acid content of the HPH was very similar to that of the HPI. However, membrane fractionation resulted in higher contents of leucine and phenylalanine in the <1 and 1–3 kDa peptides while the 3–5 and 5–10 kDa peptides had reduced contents. Fractionation also resulted in decreased levels of cysteine in the <1 and 1–3 kDa peptide fractions when compared to the other samples. Valine content was increased in the <1 and 1–3 kDa peptide fractions but remained virtually unchanged in the 3–5 and 5–10 kDa peptides, when compared to the HPI and HPH. Overall, the <1 kDa peptide fraction had the highest concentration of hydrophobic as well as aromatic amino acids except for cysteine and valine which were higher in the other peptide fractions.
Antioxidant Activities of Hemp Seed Protein Hydrolysate and Its Fractions
Radical Scavenging Activities (RSA)
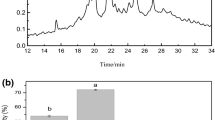
The DPPH radical is relatively stable and has been widely used to test ability of natural compounds to act as free radical scavengers or hydrogen donors as a means of evaluating their antioxidant potentials [22]. DPPH radicals are stable in methanol and show maximum absorbance at 517 nm. However, in the presence of a proton-donating substance such as an antioxidant, the DPPH radicals are scavenged leading to a reduction in absorbance. Figure 1a shows the ability of HPH and its membrane fractions to scavenge DPPH radical. The <1 kDa membrane fraction exhibited the highest RSA of 24.2% and the 5–10 kDa fraction had least activity of 18.7%. The DPPH RSA of the peptide fractions were observed to be dependent on their molecular size, with the smaller (<1 and 1–3 kDa) fractions showing higher DPPH scavenging potency than the bigger (3–5 and 5–10 kDa) peptide fractions. This result indicated that low-molecular weight (LMW) peptides possess better RSA than the high-molecular weight (HMW) peptides, which is in agreement with similar findings obtained for quinoa protein hydrolysates fractions [14] but is in contrast with results obtained for flaxseed protein-derived peptide fractions [11] and chickpea peptide fractions [8]. The unfractionated HPH was found to be a poor scavenger of DPPH (~4%) activity when compared to reduced GSH (28.2%) and the HPH fractions. Previous studies have shown that high DPPH or other radical scavenging activities of protein hydrolysates or peptide fractions are associated with high hydrophobicity [8, 23]. Current results showed that the <1 and 1–3 kDa fractions have the highest contents of hydrophobic amino acids (Table 1), which could explain the higher RSA activities of these two fractions when compared to the HPH and the higher molecular weight fractions (3–5 and 5–10 kDa). In general, the RSA of food protein hydrolysates depend on a variety of factors such as specificity of the proteases used in the protein hydrolysis, size, and amino acid composition of the peptides, and the DPPH assay conditions [11]. The RSA of the HPH fractions may be due to the presence or availability of electron donors that reduced the free radicals to more stable inert molecules. In addition, several studies have demonstrated that hydrophobic amino acids act as antioxidants by increasing the solubility of peptides in non-polar environments thereby facilitating better interaction with free radicals (such as DPPH) and terminating their reactivity [9]. Overall, the RSA of the hemp seed hydrolysates was lower than those reported for chickpea protein hydrolysates [8].

Scavenging activities of hemp seed protein hydrolysate (HPH) and its ultrafiltration fractions against DPPH radicals (a) and hydroxyl radicals (b) compared to reduced glutathione (GSH). Bars (mean ± standard deviation) with different letters have mean values that are significantly different at p < 0.05. NA no activity detected
Hydroxyl radical scavenging activity is shown in Fig. 1b. The type of free radical system utilized for antioxidant evaluation could influence experimental results; thus, the use of more than one free radical system to investigate radical-scavenging capacities of a selected antioxidant has been recommended [8, 11]. The hydroxyl radical is one of the most reactive ROS, reacting with virtually all cellular macromolecules such as proteins, polyunsaturated fatty acids, and nucleic acids to induce severe damages to cells [9] ultimately leading to aging, cancer, diabetes, and several other disease conditions [24]. Hydrogen peroxide and superoxide anion can be converted to hydroxyl radicals in vivo in the presence of metal ions or may be generated from several other radical systems leading to oxidative stress. Therefore, the scavenging of hydroxyl radical is an effective defense strategy of the human body against various diseases elicited or propagated by the ROS. The scavenging activities of HPH and its membrane fractions were compared to that of GSH (Fig. 1b). While the HPH showed no activity in scavenging hydroxyl radical, all the HPH membrane fractions exhibited similar and low hydroxyl radical scavenging activities of 15.7–23.7%. These activities were lower than that observed for GSH, which scavenged 81.7% of hydroxyl radical at the same concentration. The hemp seed peptides are poor scavengers of hydroxyl radical when compared to chickpea [8] and wheat germ [22] peptides that showed >30% activity. The results indicate that the ratio of hydroxyl radical scavenging peptides to non-scavenging peptides was very low in the HPH. However, fractionation led to increased segregation of hydroxyl radical scavenging peptides, which was reflected in the higher activity of the membrane fractions.
Metal Ion Chelating and Ferric Reducing Activities
Transition metal ions are involved in many in-vivo oxidation reactions. Ferrous ions (Fe2+) can catalyze the Haber–Weiss reaction and induce superoxide anions to form more hazardous hydroxyl radicals. These hydroxyl radicals react rapidly with adjacent biomolecules to cause severe tissue damage. Ferrous ion can also stimulate lipid peroxidation by Fenton reaction, and accelerate peroxidation by decomposing lipid hydroperoxides into hydroxyl and alkoxyl radicals capable of abstracting hydrogen and perpetuating a chain reaction of lipid peroxidation [15, 25]. Chelating agents may serve as secondary antioxidants because they reduce the redox potential, hence stabilizing the oxidized form of the metal ions. Figure 2a shows the Fe2+ chelating effects of reduced GSH, HPH, and its membrane fractions. The Fe2+ chelating ability was estimated in this study by the decrease in the absorbance of ferrozine–Fe2+ complex after the addition of the test samples. Clearly, the HPH exhibited the strongest chelating capacity (72%) which could be attributed to the additive effects of all the peptides that are responsible for the activities of the fractions. However, GSH and the <1 and 1–3 kDa HPH fractions showed low chelating activity ranging from 15.7 to 20.2% compared to the moderate activity exhibited by the 3–5 and 5–10 kDa HPH fractions, which had 29 and 38.5% Fe2+ chelating activity, respectively. Thus, the metal ion chelating activities of HPH fractions increased with the molecular weight (probably due to additive effects from constituent peptides) and the synergistic effects of the fractions were reflected in the high activity of the unfractionated hydrolysate. Fractions 3–5 and 5–10 kDa also contained the highest levels of negatively charged amino acids, which could have contributed to increased electrostatic affinity for the positively charged iron. The present results are similar to those from a previous work [5], which showed that increased peptide chain length led to higher iron chelating effects. The observed iron chelating property of HPH and its fractions may be beneficial towards the protection of cellular components against oxidative damage.

Metal chelating effects (a) and ferric reducing power (b) of hemp seed protein hydrolysate (HPH) and its fractions compared to reduced glutathione (GSH). Bars (mean ± standard deviation) with different letters have mean values that are significantly different at p < 0.05
The ferric reducing antioxidant power (FRAP) assay is used to evaluate the ability of natural antioxidants to donate electrons. Some reports have indicated that there is a direct correlation between antioxidant activities and reducing power of protein hydrolysate fractions [25, 26]. Figure 2b shows the FRAP of GSH, HPH and membrane fractions measured at 700 nm. An increase in absorbance indicates better reducing power of the test sample. The HPH and its membrane fractions exhibited low absorbance values of 0.06–0.13 compared to GSH which had highest absorbance value of 2.29. This implied that GSH had the highest reducing power when compared to HPH and its membrane fractions. However, the FRAP of the hemp seed hydrolysates is similar to values previously reported for chickpea protein hydrolysates [8]. There was an increase in reducing power of the HPH fractions with increasing peptide size (3–5 and 5–10 kDa fractions were better than <1 and 1–3 kDa fraction), which is an indication of additive effects of active groups within the peptides; long-chain peptides will contain more reducing groups than short-chain peptides. The HPH had significantly higher values than the <1 and 1–3 kDa fractions, which suggest that the presence of 3–5 and 5–10 kDa peptides contributed to the observed HPH activity. The trend showing higher FRAP of the 3–5 and 5–10 kDa fractions are similar to the trend observed for the iron chelating effects of the samples. The presence of amino acids leucine, isoleucine, histidine, methionine, tyrosine, lysine, and tryptophan could have contributed to the reducing power activity observed for the protein hydrolysates [27].
Inhibition of Linoleic Acid Oxidation
Peroxidation of fatty acids causes deleterious effects in foods by forming a complex mixture of secondary metabolites of lipid peroxides. When these affected foods are consumed, they can cause some adverse effects including toxicity to mammalian cells [8]. Lipid peroxidation proceeds via radical mediated abstraction of hydrogen atoms from methylene carbons in polyunsaturated fatty acids [28]. The antioxidant activities of GSH, HPH, and its membrane fractions against peroxidation of linoleic acid were evaluated and the results obtained after 7 days of incubation. Table 2 shows that the HPH and fractionated samples effectively inhibited linoleic acid autoxidation at varying degrees similar to the effect of GSH. After the 4th day, GSH and the peptide samples seem to gradually lose their antioxidant effects as evident by slight increases in absorbance from day 5–7. The present results are similar to previous work [22], which showed that wheat germ protein hydrolysate gradually lost its effectiveness against linoleic acid oxidation after 3 days of incubation. Overall, HPH showed the highest activity at day 7 in protecting linoleic acid against peroxidation, which may be due to the additive effects of all the active peptides present in the different fractions. The strong antioxidant effects of the peptide samples may have been due to the high levels of hydrophobic amino acids, which enhanced solubility of the peptides in the lipid phase and thus facilitating better interactions with free radical species [29]. In addition, the presence of histidine (His) in the peptides has also been reported to act against lipid peroxidation because His possess an imidazole ring in its structure, which may be involved in hydrogen donation and lipid radical trapping ability [30]. The observed rapid increase in linoleic acid oxidation in the absence of antioxidants (control) up to the 4th day is similar to previous reports [23, 31]. As the incubation proceeds beyond the 4th day, the linoleic acid is probably depleted and production of reactive oxidation products (e.g., hydroperoxides) becomes limited, which is believed to be responsible for the sharp decrease in absorbance values [31].
Conclusions
This study has shown that the HPH and its ultrafiltration fractions have effective in-vitro antioxidant properties. The ability of the peptides to scavenge hydroxyl and DPPH radicals, reduce ferric metal ions and chelate transition metal ions is indicative of their potential use for managing metabolic disorders that arise from excessive levels of ROS. Based on the results from this work, defatted hemp seed meal possesses the potential for use as raw material for the production of peptide ingredients that could be used to formulate functional foods and nutraceuticals with multifunctional bioactive properties against various free radicals that may cause oxidative stress-related diseases. However, in-vivo availability, potency, and safety must be determined before the products can be used for therapeutic purposes.
References
Yin SH, Tang CH, Cao JS, Hu EK, Wen QB, Yang XQ (2006) Effects of limited enzymatic hydrolysis with trypsin on functional properties of hemp (Cannabis sativa L.) protein isolate. Food Chem 106:1004–1013
Oomah BD, Busson M, Godfrey DV, Drover JCG (2002) Characteristics of hemp (Cannabis sativa L.) seed oil. Food Chem 76:33–43
Tang CH, Ten Z, Wang XS, Yang XQ (2006) Physicochemical and functional properties of hemp (Cannabis sativa L.) protein. J Agric Food Chem 54:8945–8950
Wang XS, Tang CH, Yang XQ, Guo WR (2008) Characterization, amino acid composition and in vitro digestibility of hemp (Cannabis sativa L.) proteins. Food Chem 107:11–18
Tang CH, Wang XS, Yang XQ (2009) Enzymatic hydrolysis of hemp (Cannabis sativa L.) protein isolate by various proteases and antioxidant properties of the resulting hydrolysates. Food Chem 114:1484–1490
Guo H, Kouzuma Y, Yonekura M (2009) Structures and properties of antioxidative peptides derived from royal jelly protein. Food Chem 113:238–245
Urso ML, Clarkson PM (2003) Oxidative stress, exercise, and antioxidant supplementation. Toxicology 189:41–45
Li Y, Jiang B, Zhang T, Mu W, Liu J (2008) Antioxidant and free radical scavenging activities of chickpea protein hydrolysate (CPH). Food Chem 106:444–450
Kim SY, Je JY, Kim SK (2007) Purification and characterization of antioxidant peptide from hoki (Johnius belengerri) frame protein by gastrointestinal digestion. J Nutr Biochem 18:31–38
Aluko RE (2008) Antihypertensive properties of plant-derived inhibitors of angiotensin I converting enzyme activity: a review. Recent Prog Med Plants 22:541–561
Udenigwe CC, Lu YL, Han CH, Hou WC, Aluko RE (2009) Flaxseed protein-derived peptide fractions: antioxidant properties and inhibition of lipopolysaccharide-induced nitric oxide production in murine macrophages. Food Chem 116:277–284
Pena-Ramos EA, Xiong YL (2004) Antioxidant activity of soy protein hydrolysates in a liposomal system. J Food Sci 67:2952–2959
Markwell MAC, Haas SM, Biebar LL, Tolbert NE (1978) A modification of the Lowry procedure to simplify protein determination in membrane and in protein samples. Anal Biochem 87:206–211
Aluko RE, Monu E (2003) Functional and bioactive properties of quinoa seed protein hydrolysates. J Food Sci 68:1254–1258
Xie Z, Huang J, Xu X, Jin Z (2008) Antioxidant activity of peptides isolated from alfalfa leaf protein hydrolysate. Food Chem 111:370–376
Zhang SB, Wang Z, Xu SY (2008) Antioxidant and antithrombotic activities of rapeseed peptides. J Am Oil Soc 85:521–527
de Avellar IG, Magalhaes MM, Silva AB, Souza LL, Leitao AC, Hermes-Lima M (2004) Reevaluating the role of 1, 10-phenanthroline in oxidative reactions involving ferrous ions and DNA damage. Biochim Biophys Acta 1675:46–53
Bidlingmeyer B, Cohen S, Tarvin T (1984) Rapid analysis of amino acids using pre-column derivatization. J Chromatogr 336:93–104
Gehrke C, Wall L, Absheer J, Kaiser F, Zumwalt R (1985) Sample preparation for chromatography of amino acids: acid hydrolysis of proteins. J Assoc Off Anal Chem 68:811–821
Landryl J, Delhaye S (1992) Simplified procedure for the determination of tryptophan of foods and feedstuffs from barytic hydrolysis. J Agric Food Chem 40:776–779
Farzamirad V, Aluko RE (2008) Angiotensin-converting enzyme inhibition and free-radical scavenging properties of cationic peptides derived from soybean protein hydrolysates. Int J Food Sci Nutr 59:428–437
Zhu KX, Zhou HM, Qian HF (2006) Antioxidant and free radical-scavenging activities of wheat germ protein hydrolysates (WGPH) prepared with alcalase. Process Biochem 41:1296–1302
Pownall TL, Udenigwe CC, Aluko RE (2010) Amino acid composition and antioxidant properties of pea seed (Pisum sativum L.) enzymatic protein hydrolysate fractions. J Agric Food Chem 58:4712–4718
Young IS, Woodside JV (2001) Antioxidants in health and disease. J Clin Pathol 54:176–186
Yun-hui C, Zhang W, Shi-ying X (2006) Antioxidant properties of wheat germ protein hydrolysates evaluated in vitro. J Cent South Univ Technol 13:160–165
Wang JS, Zhao MM, Zhao QZ, Jiang YM (2007) Antioxidant properties of papain hydrolysates of wheat gluten in different oxidation systems. Food Chem 101:1658–1663
Qian ZJ, Jung WK, Kim SK (2008) Free radical scavenging activity of novel antioxidative peptide purified from hydrolysate of bullfrog skin (Rana catesbeiana Shaw). Bioresour Technol 99:1690–1698
Rajapakse N, Mendis E, Byun HG, Kim SK (2005) Purification and in vitro antioxidative effects of giant squid muscle peptides on free radical-mediated oxidative systems. J Nutr Biochem 16:562–569
Rajapakse N, Mendis E, Jung WK, Je JY, Kim SK (2005) Purification of radical scavenging peptide from fermented mussel sauce and its antioxidant properties. Food Res Int 38:175–182
Moure A, Dominguez H, Parajo JC (2006) Antioxidant properties of ultrafiltration-recovered soy protein fractions from industrial effluents and their hydrolysates. Process Biochem 41:447–456
Jayaprakasha GK, Singh RP, Sakariah KK (2001) Antioxidant activity of grape seed (Vitis vinifera) extracts on peroxidation models in vitro. Food Chem 73:285–290
Acknowledgments
Funding for this work was provided through a Discovery grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) and an operating grant from the Advanced Foods and Materials Network of Centres of Excellence, Canada (AFMNet) to Dr. R.E. Aluko. C. C. Udenigwe is a recipient of an NSERC Alexander Graham Bell Ph.D. scholarship.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Girgih, A.T., Udenigwe, C.C. & Aluko, R.E. In Vitro Antioxidant Properties of Hemp Seed (Cannabis sativa L.) Protein Hydrolysate Fractions. J Am Oil Chem Soc 88, 381–389 (2011). https://doi.org/10.1007/s11746-010-1686-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-010-1686-7