
Abstract
Apricot kernels were roasted for various lengths of time (0–30 min) at 180 °C and changes in the oxidative stability, antioxidant capacity, color, as well as the level of tocopherols and fatty acids of the apricot kernel oil (AKO) were monitored. While the level of tocopherols decreased, the oxidative stability and antioxidant capacity of AKO increased with roasting, probably due to the formation of antioxidative Maillard reaction products (MRPs) during the roasting. Medium roasted samples (15–20 min) were found to be more resistant to oxidative deterioration. The oil from the 30-min roasted sample was more susceptible to oxidation compared to the oil from the 20-min roasted sample in most of the stability tests. Relatively shorter roasting periods (5–10 min) also led to a decrease in oxidative stability in comparison to the unroasted sample. Brownish color and antiradical activity increased with roasting and the highest values were measured in the 30 min roasted sample.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Apricot (Prunus armeniaca L.) is a stone fruit that is cultivated widely around the world. Turkey is the largest apricot producing country with an average annual production of up to 500,000 tones [1]. Apricot kernels, a by-product of apricot fruit production, are a fair source of oil and protein [2]. Apricot kernel oil (AKO), which is rich in oleic acid and γ-tocopherol, constitutes 50% of the apricot kernel weight [3]. The main utilizer of AKO is the cosmetic industry who use it as a carrier oil [4]. However, AKO can also be used for edible purposes due to its high nutritional value [5].
Oxidation is one of the leading causes of oil deterioration resulting in off flavors and rancidity [6]. Self protection against these oxidation reactions is provided by phytochemicals such as tocols, phenolic compounds and carotenoids present in vegetable oils. Maillard reaction products (MRPs) have also attracted great attention in recent years due to their antioxidative potential. Antioxidative behavior of MRPs has been reported in both model systems [6, 7] and in heated food samples [8]. High temperatures are applied to oily seeds prior to oil extraction in order to increase the oil yield, inactivate enzymes and denaturate proteins [9]. Moreover, like most of the other nuts, apricot kernels are generally consumed after being roasted [10]. During these heat processes, MRPs are formed and are passed on into the oil during extraction. The oxidative stability of rapeseed, sunflower [11], soybean [12], sesame [13], mustard, canola [9], and safflower [14] oils are enhanced by heat pretreatment prior to oil extraction. Hence, it could conceivably be hypothesized that the MRPs formed during roasting might play a role in the improvement of the oxidative stability of AKO. So far, however, no research has been reported on the effect of roasting on antioxidant capacity and oxidative stability of AKO, which is the objective in the present study.
Materials and Methods
Materials
A tocopherol standard (50 mg of α, β, γ and δ-tocopherol mixture) was purchased from Calbiochem (La Jolla, CA). A fatty acid methyl ester (FAME) mixture (37 component FAME mix) was purchased from Supelco (Bellefonte, PA). All the other chemicals and reagents for the analysis were analytical or chromatographic grades.
Sample Preparation
Apricot seeds were obtained from local markets and stored at +4 °C with their outer shells until used. Just before the experiments, the outer shells were removed manually. Kernels were crumbled into particles smaller than 1 mm by using a Waring blender and sieved to obtain apricot kernel grits.
Roasting and Oil Extraction
A 100-g sample was spread on an aluminum tray in a layer to assure a 1-cm thickness and placed in an oven set at 180 °C. Samples roasted for 5, 10, 15, 20 and 30 min, were immediately cooled to room temperature and stored at −20 °C in plastic bags. Unroasted samples were also stored under the same conditions. Oils were extracted from kernels by cold pressing using a lab-type oil press (Caselsan, Turkey) and kept in glass containers under nitrogen at −20 °C until used.
Oven Test
Five grams of oils were weighed into glass petri plates (15 mm height and 80 mm diameter) and placed in a forced-draft air oven set at 70 ± 1 °C for 22 days. At intervals, plates were removed, cooled to room temperature and peroxide (PV) and p-anisidine values (p-AV) of the oil measured.
Oxidative Stability Tests
PV and p-AV were determined according to standard methods of AOCS [15] with minor modifications. For PV measurements, an appropriate amount of oil (depending on oxidation degree, 2–3 g) was taken from the petri plates and dissolved in 25 mL of a chloroform:acetic acid mixture (2:3, v:v). Thereafter, 1 mL of saturated potassium iodide solution in water was added and the samples were shaken for 1 min. Mixtures were left in darkness for 5 min and 75 mL of distilled water was added. The liberated iodine was titrated with 0.01 N sodium thiosulphate solution using a starch solution as an indicator. The PV expressed as milliequivalents oxygen kg−1 oil.
For the p-AV test, approximately 1-g oil samples were dissolved in isooctane and the volume was made up to 25 mL with the same solvent. Five milliliters of this solution was mixed with 1 mL of 2.5 g L−1 of p-anisidine reagent (in acetic acid) and kept in the dark for 10 min. The absorbance (A S) was measured at 350 nm using a double beam spectrophotometer (Shimadzu 1700, Kyoto, Japan). A blank test (without the addition of the sample) was also performed (A B) and the p-AV was calculated using the following formula: \( p - {\text{AV}} = {\frac{{A_{S} - (A_{B} \times 1.2)}}{m}},\) where m is the mass of the oil sample in grams.
A Metrohm Rancimat apparatus, model 743 (Metrohm, Switzerland) was used for the induction period (IP) determination of oil samples [16]. For this purpose, each oil sample (4.0 ± 0.1 g) was weighed into the reaction vessel glassware. The conductimetry cells were filled with deionized water up to 90 mL. Samples were heated to 110 °C and air was passed through the heated oil at the rate of 20 L h−1. The IP was determined automatically by the device and expressed as hours.
The infrared spectra of the samples were recorded on a FT-IR (Varian 1000 Model) system with a horizontal attenuated total reflectance (ATR) apparatus and Omega CN 800 (PIKE technology) heating unit [17]. The spectrometer was equipped with a deuterated triglycine sulfate detector and purged with dry nitrogen (DuraDry, Haverhill, MA). The ATR crystal was cleaned with pure chloroform before each measurement. The oil sample (400 μL) was spread as a thin layer on heated horizontal ATR crystal (130 °C) and periodical scans (18 scans, 4 cm−1 resolution) were obtained in the spectral range of 400–4,000 cm−1 at 20 min intervals for 360 min. Induction times (IT), the time needed for dramatic increase in absorbance, were determined algebraically [17].
Antioxidant Tests
Methanolic extracts of AKO samples were prepared following the method of Tuberoso et al. [18]. A 1-g oil sample and 1 mL of methanol were mixed in Eppendorf tubes and agitated with a Vortex mixer for 1 min. Samples were centrifuged at 3,000 rpm for 5 min and the upper methanolic phase was taken. Then, 1 mL fresh methanol was added and the extraction was repeated three times, upper phases were combined and diluted to 10 mL.
The radical scavenging power (RSP) were determined by the 2,2-diphenyl-1-picrylhydrazyl (DPPH) [19] and 2,2′-azino-bis-(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) [20] methods. For the DPPH test, 100 μL methanolic extracts of the oil samples were added to a 2.9-mL methanolic solution of DPPH (25 mg L−1). After 30 min of incubation in darkness, absorbances were measured at 520 nm against methanol. ABTS radical cation (ABTS·+) solution was produced by reacting 7.0 mM ABTS stock solution with 2.45 mM (final concentration) potassium persulfate in the dark for 16 h. The resulting solution was diluted with methanol by adjusting the absorbance to 0.700 ± 0.020 at 734 nm. A diluted ABTS+ solution (2.9 mL) was added to 100 μL of methanolic oil extracts and the absorbance was measured after 6 min at 734 nm. The results of both ABTS and DPPH tests were expressed as microgram trolox equivalent g−1 oil (equivalent to 10 mL methanolic extract).
The β-carotene bleaching method was used as previously described [10]. Briefly, a 2-mL solution of β-carotene in chloroform (0.2 mg mL−1) was added to 40 mg of linoleic acid and 400 mg of Tween-20. The chloroform was evaporated under vacuum at 45 °C for 4 min, and 100 mL distilled water was added with vigorous agitation to form an emulsion. Three milliliters of this emulsion was added to each tube containing 50 μL oil methanol extract. The absorbance was measured at 470 nm, immediately, against a blank consisting of the emulsion without β-carotene. The tubes were incubated in a water bath at 50 °C for 120 min. Antioxidant activity (AA) which was expressed as the delay of the bleaching of β-carotene was determined using the following formula; \( {\text{AA = 100}}\left[ { 1 - (A_{0} - A_{\text{t}} )/(A_{0}^{O} - A_{\text{t}}^{O} )} \right],\) where A 0 and \( A_{0}^{O} \) represent the absorbance values measured at zero time of the incubation for the test sample and the control, respectively. The A t and \( A_{\rm t}^{O} \) are the absorbances measured in the test sample and the control, respectively, after incubation for 120 min.
Fatty Acid Analysis
Forty milligram of oil was methylated with 3 mL of 60 g L−1 HCl in methanol at 75–80 °C for 2 h. The FAMEs were extracted with 2 mL of hexane and dried over sodium sulfate [21]. One microliter of the FAMEs was analyzed with an Agilent 7890 series gas chromatograph (Agilent Company) equipped with a flame ionization detector and a 7683B automatic injector. A fused silica DB23 capillary column (60 m × 0.25 mm i.d., 0.25 μm film thickness; J&W Scientific, Folsom, CA) was used. The oven temperature was programmed as follows: 140 °C for 5 min, increased to 240 °C at 3 °C min−1, and kept at 240 °C for 10 min. The injector and detector temperatures were each kept at 250 °C, the carrier gas was helium, the flow rate was 30 mL min−1, and the split ratio was 1/30. FAME identification was based on retention times as compared with those of the standard FAME mixture. Results were expressed as percentage of peak area without any corrections. Fatty acid analysis was performed in triplicate for each sample, and average values were reported.
Tocopherol Analysis
Tocopherol composition of the samples was determined as described by Karabulut et al. [22]. Normal phase HPLC was used to analyze tocopherols using a ThermoFinnigan HPLC system integrated with an auto-sampler including temperature controller for the column (SpectraSystem AS3000), a degasser system (SpectraSystem SCM1000), and a quaternary gradient pump (SpectraSystem P4000) (ThermoFinnigan, San Jose, CA). The chromatographic separation was achieved with a Luna Silica column (250 mm × 4.6 mm, 5 μm; Phenomenex, Torrance, CA), and the column temperature maintained at 30 °C. Separation of tocopherols was based on isocratic elution with n-hexane:isopropanol (99:1) at 1 mL min−1. The injection volume of the samples was 20 μL. The eluate was monitored at 292 nm by using a SpectraSystem UV6000LP photodiode-array detector (ThermoFinnigan, San Jose, CA). The compounds were identified by comparing their retention times and UV spectra with authentic standards. Tocopherols were quantified based on peak areas compared with external standards. Tocopherol analysis was performed in triplicate for each sample, and average values were reported.
Color Analysis
The color of the samples was determined by measuring CIE L* (lightness), a* (redness), and b* (yellowness) values with a Minolta chroma meter (model CR-20, Minolta, Japan).
Statistical Analyses
Experimental data were evaluated using analysis of variance (ANOVA) and significant differences among the means of three replicates (P < 0.05) were determined by Duncan’s multiple range test, using the “SPSS 9.0 for Windows”.
Results and Discussion
Oxidative Stability Tests
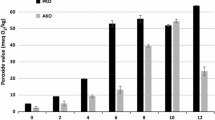
Variation in the PV of AKO samples during the oven test is shown in Fig. 1. Oils from 15 and 20 min roasted apricot kernels had the lowest (P < 0.05) PV at the end of the oven test (22nd day). However, the oils from 5- and 10-min roasted samples had a higher PV than those of unroasted and the other roasted AKO samples (Fig. 1). At the beginning of the oxidation test, oil from the 30 min roasted sample had the highest (>1.0) p-AV while the other samples including the unroasted sample had relatively low p-AV levels (Fig. 2). The higher value may be the result of the long exposure time at elevated temperature during the 30 min roasting of the apricot kernels. Roasting itself was reported to cause lipid oxidation during prolonged heating [23]. The p-AV values, from lowest to highest, were observed in samples roasted 15, 20, 0, 10, 30 and 5 min (Fig. 2). The PV and p-AVs are commonly used to estimate degree of oxidative deterioration in heated oils. The former is the measure of primary and the later is of secondary oxidation products [23]. Due to the instability of primary oxidation products in advance stages of the oxidation, measuring secondary oxidation products along with the peroxides is critical [9]. In agreement with our findings, oils obtained from roasted seeds were found to contain lower primary [14] and secondary [9] oxidation products compared to the oils from unroasted seeds.

Peroxide value (PV) versus oven oxidation time of the oils obtained from unroasted and roasted apricot kernels

p-Anisidine value (p-AV) versus oven oxidation time of the oils obtained from unroasted and roasted apricot kernels
Since AKO is rich in oleic acid, a temperature of 110 °C was used instead of the commonly used temperature of 98 °C in the Rancimat test in order to shorten the IP. This relatively higher temperature could reduce the solubility of oxygen in oil but we applied an air stream at 20 L h−1 to overcome this problem [16]. Oil from the 5-min roasted sample had the shortest Rancimat IP of 19.1 h, and the 19.7 h IP observed in oil from unroasted samples were not statistically different (P > 0.05). Gradually increasing IPs were measured in oils from the 10, 15 and 20 min roasted samples, which had IPs of 20.7, 23.0, 24.5 h, respectively (Fig. 3). Although a slight decrease in IP was observed for the 30-min roasted sample with respect to the 20-min roasted sample, it was not statistically significant (P > 0.05). The Rancimat test is a powerful tool fot estimating the oxidative stability of oils. It is generally applied along with the Schaal oven test in order to assess the oil oxidative stability at both mild and elevated temperatures [24]. Higher concentrations of antioxidants and/or lower rates of polyunsaturated fatty acids cause elongation in the Rancimat IP. There are few studies in which oils from roasted seeds have been tested for their oxidative stability by means of a Rancimat test. In one study, which was compatible with our findings, higher Rancimat IPs were reported for the oils obtained from roasted rapeseed and sunflower compared to the oils obtained from unroasted seeds [11].

Rancimat induction period (IP) of the oils obtained from apricot kernels roasted for different durations. Different letters on bars indicate statistically significant differences between the mean (P < 0.05)
FT-IR spectroscopy is a recent technique for detecting and quantifying functional groups that arise during the oxidative degradation of lipids [25]. The ATR crystal allows one to work with small oil quantities and eliminate the time consuming KBr disc preparation step. Moreover, due to the open surface to the air, the ATR crystal is very suitable for real-time oxidation experiments [17]. In the present study, formation of different oxidation products was monitored by their characteristic wavenumber regions. Spectral regions were given instead of strict wavenumber values due to the changeable absorbance maxima bands observed for a certain functional group throughout the oxidation period [12]. The FT-IR spectrum of unroasted AKO, which was subjected to oxidation, is shown on Fig. 4. Several spectral data were collected throughout oxidation experiment but only non-oxidized, medium oxidized and fully oxidized samples’ spectra are shown in Fig. 4 for clarity. During oxidation, absorbance of some groups indicated by arrows increased continuously while others decreased in the course of the oxidation period (Fig. 4). Table 1 summarizes the wavenumber regions, ITs and the chemical groups that are probably responsible for the increase/decrease in absorbance. There were increases and decreases in some of other wavenumber regions but we evaluated only the regions that are known to be due to certain oil oxidation products. The wavenumber region of 700–725 cm−1 belongs to cis double bonds in unsaturated fatty acids [26]. During oxidation, due to the loss of cis double bonds, the absorbance intensity in this region decreases, as we observed in our study. An increase in absorbance that we observed at the region of 960–975 cm−1 was probably the result of the formation of trans double bonds that arose in the course of heat induced oxidation [27]. Secondary oxidation products such as aldehydes and ketones are known to have absorption in the 880–890 cm−1 [25] and 1,690–1,700 cm−1 [25, 28] regions. Thus, the absorbance increase we observed near these regions could be attributed to the accumulation of secondary oil oxidation products.

FT-IR spectrum of unroasted and nonoxidized apricot kernel oil. Arrows indicate the regions where absorbance increases/decreases occur in the course of the oxidation process
A reduction in the absorbance near the 3,008–3,010 cm−1 wavenumber region was possibly due to the disappearance of cis double bonds as in the case of the 700–725 cm−1 band [27]. The absorbance increase near the 3,400–3,600 cm−1 region was reported to be caused by the formation of hydroperoxides and alcohols [25, 28]. Generally it is accepted that 3,444 cm−1 is a characteristic band for hydroperoxides.
Data obtained from the PV, p-AV, Rancimat and FT-IR tests indicate that oxidation of AKO can be retarded by kernel roasting before oil extraction. However, ITs measured for different spectral regions was slightly different. In the hydroperoxide region (3,400–3,800 cm−1), a decrease in IT was observed for 5- and 10-min roasted samples compared to the unroasted sample. Similar patterns were observed for 700–725, 960–975 and 3,008–3,010 cm−1 wave number regions. In comparison with unroasted sample, higher or equal ITs were observed for 5- and 10-min roasted samples at 880–890, 1,690–1,700 and 1,740–1,745 cm−1 wave number regions. The samples that roasted for 15 and 20 min, had higher ITs in all spectral regions compared to the unroasted sample and 5- and 10-min roasted samples. However, the sample roasted for 30 min showed the lowest IT in all the spectral regions in comparison to all AKO samples, including an unroasted sample.
Fatty Acid Composition
Oleic acid (18:1) was found to be the primary fatty acid in unroasted AKO (68.69%). Linoleic acid (18:2) and palmitic acid (16:0) contents were found to be 23.19 and 6.00% of the total fatty acids, respectively (Table 2). Other fatty acids were found in relatively lower levels. According to a recent report, oils of different apricot kernel varieties were found to average 70.83% oleic acid, 21.96% linoleic acid and 4.92% palmitic acid [3]. These results are in agreement with our findings. As reported for other oil seeds [29], roasting did not cause changes in fatty acid composition of AKO, which was confirmed by statistical analysis (P < 0.05).
Color Analysis
The color of oils obtained from unroasted and roasted apricot kernels were compared. A clear variation in all color parameters (L*, a*, b*) with roasting was observed (Table 2). Brown-colored compounds which formed during heat processes were probably the cause of these variations. Maillard reactions and sugar caramelization are two of the main causes of browning in food systems [23]. Several colored and colorless intermediates and end products are formed during Maillard reactions. It is well known that some MRPs have an antioxidant capacity at certain levels [6, 7, 9]. However, it was reported that all MRPs with antioxidant activity do not necessarily have a dark color [10]. Thus, although measurement of the browning is a powerful tool to estimate the degree of roasting, a linear relationship between color values and antioxidant capacity or oxidative stability may not always be expected.
Antioxidant Tests
The antioxidant capacity of methanolic extracts of AKO increased with increasing roasting time for both ABTS and DPPH tests (Table 2). In both tests, the unroasted sample had the lowest RSP, although the increase was not statistically significant for 5 min in ABTS and for 5 and 10 min in the DPPH test. A dramatic increase in RSP was observed for the extracts from samples roasted 15 min and longer in both tests. However, in the β-carotene linoleate assay, the increase in protection of β-carotene from bleaching was attenuated after 20 min of roasting but slightly decreased for the 30-min roasted sample. The β-carotene linoleate model system is a measure of the extracts’ oxygen trapping activity; however, DPPH and ABTS tests involve electron transfer from antioxidant to synthetic radicals [30]. Thus, depending on the composition and the nature of the individual antioxidant molecules in the extracts, observing different results from these tests is normal. The findings obtained from the β-carotene linoleate test were supportive of the PV, p-AV, FT-IR tests and the previous report in which roasting caused an increase in antioxidant capacity to a degree but further roasting caused a reverse effect [10].
Tocopherol Content
Tocopherol contents of unroasted and roasted AKOs are shown in Table 2. The gamma isomer was found to be the principle tocopherol isomer, while α and δ tocopherols were present in relatively small quantities and β-tocopherol was absent in apricot kernel oil. These findings are in agreement with a previous report [3]. Compared to unroasted sample, a significant reduction (P < 0.05) was observed for γ-tocopherol in roasted samples.The content of γ-tocopherol for the unroasted sample was measured as 452 mg kg−1 oil. Tocopherol contents of 5-, 10-, 15-, 20- and 30-min roasted samples were quantified as being 407, 427, 379, 377 and 375 mg kg−1 oil, respectively. The levels of α and δ-tocopherol isomers were not significantly (P > 0.05) effected by roasting. Tocopherol depletion could be the result of thermal degradation during roasting. A similar decrease in tocopherol content was reported for sesame seeds roasted at 160–250 °C for different durations [29]. In contrast, tocopherols have been reported to increase with roasting due to the cell damage and increasing extractability of tocopherols [14]. Thus, our findings support that roasting causes a decrease in tocopherol content.
In the light of all the findings listed above, we can postulate that antioxidant capacity and oxidative stability of AKO was improved by roasting in general. Although a clear decrease in tocopherols, the primary antioxidant molecules of vegetable oils [31] roasting longer than 15 min prior to oil extraction caused an obvious increase in oxidative stability and antioxidant capacity measured by the Rancimat test and ABTS/DPPH radical scavenging assays, respectively. Roasting was reported to improve oxidative resistance of some of other vegetable oils [11, 13, 14]. This improved stability and activity is most probably provided by the MRPs formed during roasting, which are the products of the reactions that took place between reducing sugars and amines at elevated temperatures [7]. It was shown that MRPs obtained from model systems could also retard the oxidative deterioration of oils [6].
The results of the PV, p-AV, FT-IR IT and β-carotene bleaching tests showed that a long roasting time (30 min) did not enhance oxidative stability and antioxidant capacity more than that of 20 min. Thus, extending roasting time beyond 20 min is not necessary. During roasting, degradation of naturally occurring antioxidants and formation of antioxidant MRPs occurs together. Under severe roasting conditions, the degradation rate might have been higher than the formation of MRPs and the total antioxidant capacity could be reduced [32].
On the other hand, the oxidative stability of 5- and 10-min roasted samples were found to be alike or lower than that of the unroasted sample. This may be the result of the decrease in tocopherol content caused by roasting. Oil-degrading enzymes are known to cause lipid oxidation during storage [11]. The lower oxidative stability of 5- and 10-min roasted samples could also be the result of the remaining activity of these enzymes. Meanwhile, since an adequate roasting effect was not achieved, antioxidative MRPs might not have formed at this degree of roasting. The pro-oxidant potential of early MRPs or intermediates could also have negatively affected the oxidative resistance of 5- and 10-min roasted AKO samples. A slight decrease in the antioxidant capacity during the early stages of the Maillard reactions have been observed by some of other researchers [33, 34].
Conclusions
Oils obtained from medium roasted kernels were found to be more resistant to oxidative deterioration. However, compared to an unroasted sample, shorter (5–10 min) or longer (30 min) roasting periods caused a counter effect and lower oxidative stability values were measured. In contrast to oxidative stability test results, the level of γ-tocopherol, the primary tocopherol isomer of AKO, was lowered by roasting. On account of this, it could be presumed that the disappearance of tocopherols was compensated for by the MRPs that appeared during the heat pretreatment and oxidative stability of AKO was increased to a degree. These findings were confirmed by antioxidant tests in which DPPH and ABTS•+ radicals were scavenged better by the methanolic extracts of roasted oils compared to an unroasted sample. According to the results of this study, we can state that the shelf life of oily nuts and seeds could be improved by an appropriate roasting. Further studies could be devoted to clarify the molecular structure of the individual MRPs that have a role in the protection of oils against oxidative deterioration.
References
FAO, Statistical Databases, http://faostat.fao.org/faostat (accessed on March 2009)
Gomez E, Burgos L, Soriano C, Mar J (1998) Amygdalin content in the seeds of several apricot cultivars. J Sci Food Agric 77:184–186
Turan S, Topcu A, Karabulut I, Vural H, Hayaloglu AA (2007) Fatty acid, triacylglycerol, phytosterol, and tocopherol variations in kernel oil of Malatya apricots from Turkey. J Agric Food Chem 55:10787–10794
Silem A, Günter HO, Einfeldt J, Boualia A (2006) The occurrence of mass transport processes during the leaching of amygdalin from bitter apricot kernels: detoxification and flavour improvement. Intl J Food Sci Tech 41:201–213
Gandhi VM, Mulky MJ, Mukerji B, Iyer VJ, Cherian KJ (1997) Safety evaluation of wild apricot oil. Food Chem Toxicol 35:583–587
Wagner KH, Derkits S, Herr M, Schuh W, Elmadfa I (2002) Antioxidative potential of melanoidins isolated from a roasted glucose–glycine model. Food Chem 78:375–382
Morales FJ, Perez SJ (2001) Free radical scavenging capacity of Maillard reaction products as related to colour and fluorescence. Food Chem 72:119–125
Summa CA, Calle BDL, Brohee M, Stadler RH, Anklam E (2007) Impact of the roasting degree of coffee on the in vitro radical scavenging capacity and content of acrylamide. LWT-Food Sci Technol 40:1849–1854
Wijesundera C, Ceccato C, Fagan P, Shen Z (2008) Seed roasting improves the oxidative stability of canola (B. napus) and mustard (B. juncea) seed oils. Eur J Lipid Sci Technol 110:360–367
Durmaz G, Alpaslan M (2007) Antioxidant properties of roasted apricot (Prunus armeniaca L.) kernel. Food Chem 100:1177–1181
Veldsink JW, Muuse BG, Meijer MMT, Cuperus FP, van de Sande RLKM, van Putte KPAM (1999) Heat pretreatment of oilseeds: effect on oil quality. Lipid-Fett 101:244–248
Jung MY, Bock JY, Back SO, Lee TK, Kim JH (1997) Pyrazine contents and oxidative stabilities of roasted soybean oils. Food Chem 60:95–102
Abou-Gharbia HA, Shehata AAY, Shahidi F (2000) Effect of processing on oxidative stability and lipid classes of sesame oil. Food Res Int 33:331–340
Lee YC, Oh HI, Chang J, Kim IH (2004) Chemical composition and oxidative stability of safflower oil prepared from safflower seed roasted with different temperatures. Food Chem 84:1–6
Firestone D (ed) (1997) Official methods and recommended practices of the American Oil Chemists Society, 5th edn. AOCS press, USA
Mateos R, Uceda M, Aguilera MP (2006) Relationship of Rancimat method values at frying temperatures for virgin olive oils. Eur Food Res Technol 223:246–252
Russin TA, Boye JI, Pham HM, Arcand Y (2006) Antioxidant properties of genistein in a model edible oil system. J Food Sci 71:395–399
Tuberoso CIG, Kowalczyk A, Sarritzu E, Cabras P (2007) Determination of antioxidant compounds and antioxidant activity in commercial oilseeds for food use. Food Chem 103:1494–1501
Bondet V, Brand-Williams W, Berset C (1997) Kinetics and mechanism of antioxidant action using the DPPH free radical method. Lebensm Wiss Technol 30:609–615
Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Rad Biol Med 26:1231–1237
Jennings BH, Akoh CC (1999) Enzymatic modification of triacylglycerols of high eicosapentaenoic and docosahexaenoic acids content to produce structured lipids. J Am Oil Chem Soc 76:1133–1137
Karabulut I, Topcu A, Yorulmaz A, Tekin A, Ozay DS (2005) Effects of industrial refining process on some properties of hazelnut oil. Eur J Lipid Sci Technol 107:476–480
Anjum F, Anwar F, Jamil A, Iqbal M (2006) Microwave roasting effects on the physico-chemical composition and oxidative stability of sunflower seed oil. J Am Oil Chem Soc 83:777–784
Farhoosh R, Moosavi SMR (2007) Rancimat test for the assessment of used frying oils quality. J Food Lipids 14:263–271
Guillen MD, Cabo N (1999) Usefulness of the frequency data of the Fourier transform infrared spectra to evaluate the degree of edible oils. J Agric Food Chem 47:709–719
Muik B, Lendl B, Molina-Diaz A, Valcarel M, Ayora-Cañada MJ (2007) Two-dimensional correlation spectroscopy and multivariate curve resolution for the study of lipid oxidation in edible oils monitored by FTIR and FT-Raman spectroscopy. Anal Chim Acta 593:54–67
Russin TA, van de Voort FR, Sedman J (2003) Novel method for rapid monitoring of lipid oxidation by FTIR spectroscopy using disposable IR cards. J Am Oil Chem Soc 80:635–641
Dubois J, van de Voort FR, Sedman J, Ismail AA, Ramaswamy HR (1996) Quantitative Fourier transform infrared analysis for anisidine value and aldehydes in thermally stressed oils. J Am Oil Chem Soc 73:787–794
Yoshida H, Takagi S (1997) Effects of seed roasting temperature and time on the quality characteristics of sesame (Sesamum indicum) oil. J Sci Food Agric 75:19–26
Prior RL, Wu X, Schaich K (2005) Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements. J Agric Food Chem 53:4290–4302
Kamal-Eldin A, Appelqvist LA (1996) The chemistry and antioxidant properties of tocopherols and tocotrienols. Lipids 19:671–701
Morales FJ, Martin S, Açar ÖÇ, Arribas-Lorenzo G, Gökmen V (2009) Antioxidant activity of cookies and its relationship with heat-processing contaminants: a risk/benefit approach. Eur Food Res Technol 228:345–354
Anese M, Manzocco L, Nicoli MC, Lerici CR (1999) Antioxidant properties of tomato juice as affected by heating. J Sci Food Agric 79:750–754
Nicoli MC, Anese M, Parpinel M (1999) Influence of processing on the antioxidant properties of fruit and vegetables. Trends Food Sci Tech 10:94–100
Acknowledgments
This study was supported by the Inonu University Scientific Research Center (Project No: 2004/10). The authors are grateful to the Malatya Governorships’ Apricot Research, Development and Introduction Foundation for financial support and Fiskobirlik S.S. for laboratory assistance.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Durmaz, G., Karabulut, İ., Topçu, A. et al. Roasting-Related Changes in Oxidative Stability and Antioxidant Capacity of Apricot Kernel Oil. J Am Oil Chem Soc 87, 401–409 (2010). https://doi.org/10.1007/s11746-009-1518-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-009-1518-9