
Abstract
Sphingosine-1-phosphate (S1P) is an amphiphilic signaling molecule, which is enriched in functional high density lipoprotein (HDL) and shows arterial protection. The distribution of S1P is changed with increased plasma phospholipid transfer protein (PLTP) activity and impaired HDL function in patients with coronary heart diseases. Therefore, we hypothesized that PLTP might transfer S1P among cells or lipoproteins. We found that plasma S1P contents were decreased by 60.1 % in PLTP knockout mice (PLTP−/−, N = 5) compared with their wild type littermates (WT, N = 5) (151.70 ± 38.59 vs. 379.32 ± 59.90 nmol/l, P<0.01). S1P content in HDL fraction (HDL-S1P) from PLTP−/− was decreased by 64.7 % compared with WT (49.36 ± 1.49 vs. 139.76 ± 2.94 nmol/l, P<0.01). The results of the S1P transfer assay indicated that PLTP could facilitate S1P transport from erythrocytes to HDL at 37 °C in D-Hanks buffer. Plasma content of apolipoprotein M, a specific adaptor of S1P, was not changed in PLTP−/− compared with WT. Therefore, we concluded that PLTP was a key factor to maintain plasma HDL-S1P, and PLTP deficiency could decrease the S1P content in plasma lipoproteins, which involves its capability of transferring S1P from erythrocyte to HDL.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The primary cause of death in humans is atherosclerotic disease, including coronary heart disease (CHD) and cerebral stroke [1]. High density lipoprotein (HDL) is the most important factor of anti-atherosclerosis [2]. To date, more and more researchers have proved that the significance of higher levels of HDL is considered another critical point in preventing atherosclerosis [2, 3]. The components on HDL particles determine the anti-atherosclerotic function of HDL [4]. Apolipoproteins, enzymes, and certain important lipid molecules contribute to the anti-atherosclerotic functions of HDL [4].
Sphingosine-1-phosphate (S1P) is an active signaling molecule which shows arterial protective effects via its specific receptors [5]. More than 50 % of plasma S1P is concentrated in HDL [5], and the metabolism of S1P indicates that the cellular sources of plasma S1P includes erythrocytes, endothelial cells, platelets, and mast cells [5–8]. Moreover, accepted evidence demonstrated that HDL preserves S1P via apolipoprotein M, a specific adaptor of S1P in plasma [9]. However, the transport mechanisms of secreted S1P from cells to HDL, and whether S1P distribution affected by the plasma proteins which involve HDL metabolism have not been elucidated.
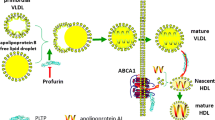
Phospholipid transfer protein (PLTP) is a multifunctional protein widely expressed and secreted by hepatocytes, adipocytes, and macrophages. Phospholipids, free cholesterol, and other amphiphilic lipid molecules can be transferred by PLTP from TAG rich lipoproteins (TRL) to HDL and vice versa [10]. Furthermore, in CHD patients, increased PLTP activity and decreased HDL levels were well documented [11, 12]. On the other hand, Sattler and her coworkers reported that plasma S1P was increased in patients with coronary artery disease or myocardial infarction, indicating that the alternation of S1P level or distribution showed a close relationship with the severity in patients with atherosclerotic diseases [13]. In addition, S1P is the phosphorylation metabolite of sphingosine, suggesting that its structure characteristics are similar to other amphipathic molecules which are transferred by PLTP [14]. Therefore, to elucidate the possible mechanism, we hypothesized that PLTP could change the content of S1P in HDL or other lipoprotein particles via its transfer activity. Our findings in the present study showed that the S1P content of HDL from PLTP knockout mice (PLTP−/−) was significantly reduced compared with their wild type (WT) littermates.
Materials and Methods
Animals
PLTP−/− were presented by Dr. Xiancheng Jiang and all animals were on a homogenous C57BL/6 background (nine generation backcrosses). PLTP−/− and WT used in this study were the offspring littermates of PLTP heterozygous mice. The mice were fed chow diet (KEAOXIELI FEED. Co. LTD, Beijing, China) and water ad libitum. Experimental animals were housed in a temperature- and humidity-controlled room with a 12/12 h light–dark cycle. This study was approved by the Laboratory Animal Care Committee of Taishan Medical University, and all animal experiments were conducted in accordance with the Guidelines for Care and Use of Laboratory Animals at the Taishan Medical University.
Antibodies and Materials
Antibodies against apolipoprotein M (apoM) were purchased from Cell Signaling Technology (Danvers, MA, USA). Complete protease inhibitor cocktail tablets were purchased from Roche (Schweiz, Germany). D-erythro-S1P, centrifugal filters (molecular weight cutoff ≤3,000 Dalton), analytical grade formic acid, and ammonium formate were obtained from Sigma-Aldrich Inc. (St. Louis, MO, USA). Internal standard D-erythro-C17-sphingosine-1-phosphate (C17-S1P) was purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Methanol and ethanol were products from SK Chemicals (Ulsan, Korea). Lipid quantification assay kits were purchased from BioSino (Beijing, China). Protein quantification kits, the dialysis membranes (molecular weight cutoff ≤8,000 Dalton), and D-Hanks buffer were provided by Solarbio (Shanghai, China).
Plasma Isolation and Lipoprotein Preparation
At 10–11 weeks of age, the mice were killed and fasting blood or organs were harvested. Samples of 0.6 ml plasma from female WT (N = 5) or female PLTP−/− (N = 5) were loaded onto ultracentrifugation for lipoprotein isolation following the methods we reported previously [15].
Preparation of S1P Standard
The stock solution of the S1P standard with a concentration of 0.1 mg/ml was prepared in ethanol, immediately aliquoted, and then stored at −20 °C. The working solutions (50–1,000 ng/ml) were prepared via dilution of the stock solution.
Biological Sample Preparation and Lipid Extraction
For lipid extraction, 20 μl plasma samples including 10 μl internal standard C17-S1P (400 ng/ml) were thoroughly mixed with methanol, followed by sonication for 10 min, and then centrifuged at 11,000 × g for 10 min [16]. The supernatants were transferred to glass vials prior to LC–MS/MS analysis. 10 μl internal standard solution (400 ng/ml) and methanol were prepared to obtain calibration standards from 5 to 100 ng/ml with internal standard at the final concentration of 40 ng/ml.
LC–MS/MS Analysis
Chromatographic separations were carried out with a linear elution using a Waters Symmetry® C18 column (3.5 μm, 2.1 mm i.d. × 100 mm) with a Waters C18 guard column (3.5 μm, 2.1 mm i.d. × 10 mm). The mobile phase was composed of 2 mM ammonium formate and 0.1 % formic acid in 90 % methanol. The injection volume was 5 μl, and the flow rate was 0.2 ml/min.
The mass spectrometer was operated in the positive ion mode with an ionspray voltage of 5,500 V at 550 °C and supplied by auxiliary gas 55 psi. Nebulizer gas was set at 55 psi, curtain gas at 10 psi and collision gas at medium. Precursor to product ion transitions of m/z 380.2 → 264.1 for S1P (collision energy 29.0 V) and m/z 366.3 → 250.3 for C17-S1P (collision energy 19.9 V) was used as a quantifier for multiple reaction monitoring (MRM) with a dwell time of 100 ms. Quantification was performed with Analyst Software 1.6 by the standard-addition method. Ratios of S1P peak area and internal standard peak area were plotted against ratios of nominal S1P concentration and internal standard concentration and calibration curve was then used to calculate the concentration of S1P in samples.
S1P Transferring Assay
To determine the transferring effect of S1P by PLTP, we developed a donor–acceptor transferring system. Briefly, fresh isolated mouse red blood cells (RBC) from PLTP−/− were washed with ice-cold D-Hanks buffer and centrifuged in 600 × g at 4 °C for 5 min, supernatant was removed. The washing step was repeated once to remove trace plasma. The cell density of RBC suspension was adjusted in the range of 107–108 per milliliter as S1P donor (the avenge S1P content in 20 μl donor was 900.67 ± 6.68 pg/ml). HDL from PLTP−/− (the avenge S1P content was 176.39 ± 24.41 pg/ml) or WT (the avenge S1P content was 325.79 ± 80.28 pg/ml) isolated by ultracentrifugation (density range 1.063–1.21 g/ml) was dialyzed in phosphate buffer solution (pH = 7.4) to remove NaBr in lipoprotein fraction. The protein concentration of HDL was determined as acceptor loading reference. Fresh plasma from PLTP−/− and WT were employed as PLTP source for the transferring assay and designated as “negative control” or “PLTP group”, respectively. D-Hanks buffer combined with donor and acceptor was designated as “blank control” of transferring assay. The 20 μl donor, 50 μl acceptor, and 2 μl PLTP source were combined in Eppendorf tubes and mixed gently. The total volume of the mixture was brought to 100 μl by D-Hanks buffer. After incubating at 37 °C for 60 min, the mixture was centrifuged in 300 × g at 4 °C for 2 min to isolate supernatant HDL and RBC pellet. S1P content in supernatant and pellet was determined. To clarify the S1P binding form in transferring assay, we carried out a pretest, and the supernatant was concentrated with centrifugal filter (cutoff molecular weight 3,000 Dalton), S1P contents in the flow-through fraction and the preserved fraction were determined. The pretest results showed that no S1P was detected in flow-through fraction, indicating that most of S1P was presence in a protein binding form (Data not shown). All steps excepted incubation was performed on ice or at 4 °C and the RBC transferring was carried out using cut-off tips.
Protein Isolation, Electrophoresis, and Western Blot
Proteins were isolated using lysis buffer with complete protease inhibitor cocktail tablets. Plasma or lipoprotein samples (2μg protein each well) used in SDS-PAGE and western blot analysis were described previously [17]. Densitometry analysis was conducted using Image-Pro Plus software version 6.0 (Media Cybernetics Corp, Bethesda, MD, USA).
Data Analysis
All the bioassay results were expressed as the mean ± standard deviation (SD). To compare the means between two groups, student's t test was performed. The differences were considered to be significant at a P value < 0.05.
Results
PLTP Deficiency Showed a Lower Plasma Total Cholesterol and HDL-C
At 10 weeks, the plasma from PLTP−/− or WT was harvested and the lipid level was determined. As shown in Table 1, plasma total cholesterol (TC) and HDL cholesterol (HDL-C) were significantly decreased in PLTP−/− by 49.3 and 55.4 % (TC, 1.31 ± 0.46 vs. 2.56 ± 0.39 mmol/l; HDL-C, 0.99 ± 0.15 vs. 2.21 ± 0.38 mmol/l, P<0.01) compared with the WT littermates. The data were consistent with previous reports, indicating that the lack of PLTP expression could dramatically lower the plasma TC and HDL-C in mice [10, 18]. Other information about the genotype and phenotype of PLTP−/− and WT littermates was mentioned in supplementary Fig. 1.

Both plasma S1P contents and cholesterol normalized S1P contents were decreased in PLTP−/−. Samples of 30 μl fresh plasma including 30 μl internal control C17-S1P (400 ng/ml) were thoroughly mixed with methanol, followed by sonication for 10 min, and then centrifuged at 11,000 × g for 10 min. The supernatants were transferred to glass vials prior to LC–MS/MS analysis. a Plasma S1P content in female PLTP−/− mice and its female WT littermates. b Total cholesterol normalized plasma S1P content of a female PLTP−/− and female WT littermates. Data were expressed as the mean ± SD (N = 5)
Both S1P Contents and Cholesterol Normalized S1P Contents were Decreased in PLTP−/− Plasma
More than 60 % of plasma S1P was enriched in lipoprotein performed its effects in cardiovascular system via S1P receptors [19]. Thus, TC and HDL-C level alternation should definitely affect plasma S1P content. As shown in Fig. 1a, plasma S1P was decreased in PLTP−/− compared with WT littermates by 60.1 % (151.70 ± 38.59 vs. 379.32 ± 59.90 nmol/l, P = 0.003). To understand the S1P alternation in lipoprotein, TC normalized plasma S1P content was displayed with the ratio of S1P content and total cholesterol. As shown in Fig. 1b, no difference was observed between WT and PLTP−/− (137.01 ± 23.10 vs. 138.15 ± 23.13 nmol S1P/mmol cholesterol, P = 0.0.918). The results suggested that the decreased plasma S1P in PLTP−/− might be closely related with lipoprotein S1P content.
PLTP Deficiency Dramatically Decreased S1P Content in HDL
To clarify the effects of PLTP deficiency on S1P distribution in lipoproteins, we isolated lipoproteins from PLTP−/− and WT for S1P content determination (see supplementary Fig. 2 for details of lipoprotein isolation). As shown in Fig. 2a, the S1P content in HDL fraction (HDL-S1P) and lipoprotein deficient plasma (LDP) fraction from PLTP−/− was dramatically reduced compared with WT by 64.7 and 69.5 %, respectively (HDL, 49.36 ± 1.49 vs. 139.76 ± 2.94 nmol/l, P<0.01; LDP 37.22 ± 6.37 vs. 121.95 ± 3.64 nmol/l, P < 0.01). After protein normalization, HDL-S1P from PLTP−/− was decreased significantly compared to WT by 46.5 % (Fig. 2b, 182.73 ± 5.50 vs. 341.95 ± 7.21 pmol/mg protein, P < 0.01). We also found that S1P in LDL or VLDL fractions was decreased; however, the S1P content in non-HDL was very minor in mice lipoproteins (Fig. 2c), indicating that the HDL-S1P declination was the main cause of low plasma S1P in PLTP−/−.

S1P content in lipoproteins was affected by PLTP expression. Pooled 0.6 ml plasma from PLTP−/− and their wild type littermates was loaded for lipoprotein isolation with series density ultracentrifugation (see supplementary Fig. 2). A 30 μl ultracentrifugation fraction including 10 μl internal standard C17-S1P (400 ng/ml) were thoroughly mixed with methanol, followed by sonication for 10 min, and then centrifuged at 11,000 × g for 10 min. The supernatants were transferred to glass vials prior to LC–MS/MS analysis. a S1P content in ultracentrifugation fraction. b Potein-normalized S1P content in lipoprotein. c Protein-normalized S1P percentage comparison. Data were expressed as the mean ± SD (N = 4). *P < 0.05, **P < 0.01, NS no significance
PLTP Transferred S1P from RBC to HDL
It is well known that the primary function in plasma of PLTP is mediating phospholipid exchange between lipoproteins or cells [20]. S1P is a phosphorylated sphingosine molecule, and its major source in plasma is erythrocytes [21]. Our data indicated that PLTP deficiency could decrease lipoprotein S1P content. To clarify the mechanism of lowered S1P content in PLTP−/− HDL-S1P, we performed a S1P transferring assay of PLTP. As shown in Fig. 3a, WT plasma increased HDL-S1P by 27.1 % (673.37 ± 21.38 vs. 491.24 ± 5.42 pmol/l, P = 0.020) compared with PLTP−/− plasma, indicated that plasma derived PLTP might transfer S1P from RBC to HDL. However, we noticed that S1P content was also increased in negative controls compared to blanks (491.24 ± 5.42 vs. 374.88 ± 7.05 pmol/l, P = 0.010), suggesting that HDL from WT might not be a suitable acceptor because PLTP is difficult to remove thoroughly by ultracentrifugation, and the trace PLTP in WT HDL could disturb the transfer assay. Therefore, to eliminate the PLTP disturbance in transferring, we used PLTP−/− HDL as the acceptor and found that WT plasma could increase HDL-S1P by 62.2 % (112.19 ± 1.08 vs. 42.41 ± 6.51 pmol/l, P = 0.018) while no S1P content difference was observed between blank and negative control (Fig. 3b, 56.31 ± 1.46 vs. 42.41 ± 6.51 pmol/l, P = 0.094). The results indicated that PLTP could transfer S1P from RBC to HDL.

PLTP transferred S1P from RBC to HDL. Fresh isolated mouse red blood cells (RBC) from PLTP−/− were used as the S1P donor. The dialyzed mouse HDL was used as the S1P acceptor in this transfer assay. Fresh plasma from PLTP−/− or WT was employed as PLTP source of transfer assay. The 20 μl donor, 50 μl acceptor, and 2 μl PLTP source were combined in an Eppendorf tube and mixed gently, and the total volume was brought to 100 μl by ice-cold D-Hanks buffer. The transfer assay mixture was incubated at 37 °C for 60 min. After incubation, the mixture was centrifuged in 300 × g at 4 °C for 2 min to isolate supernatant HDL and RBC pellet. S1P content in supernatant and pellet was determined. a The transfer assay results employing the dialyzed WT HDL as S1P donor. b The transfer assay results employing the dialyzed PLTP−/− HDL as S1P donor. Data were expressed as the mean ± SD (n = 3)
ApoM Content in Plasma and Lipoprotein was not Affected by PLTP Expression
S1P binds to HDL via its specific adaptive protein, apolipoprotein M [9]. To understand whether the content of apoM was affected by PLTP expression, apoM content in plasma and lipoproteins were detected by western blot. As shown in Fig. 4a and b, no difference of apoM in plasma or HDL was observed from PLTP−/− compared with WT. Furthermore, apoM shift was observed in many gene modified mouse models; therefore, we determined apoM in non-HDL fraction isolated by ultracentrifugation [22]. As shown in Fig. 4c, no apoM was detected in non-HDL of PLTP−/−, indicating that the apoM distribution in plasma and lipoprotein was not affected by PLTP deficiency.

ApoM content in plasma and lipoprotein was not affected by PLTP expression. Samples of 2 μg plasma or lipoprotein from wild type or PLTP−/− mice was mixed with protein loading buffer and conducted with heat denaturation for western blot analysis; all experiments were carried out for at least three times and the preventative results were shown. a Upper panel, plasma apoM contents of 10-week-old female PLTP−/− mice and female WT littermates (N = 4), lower panel, density analysis of apoM signal of western blot. b Upper panel, HDL apoM contents of 10-week-old female PLTP−/− mice and female WT littermates (N = 3), lower panel, density analysis of apoM signal of western blot. c ApoM content in non-HDL (combined VLDL and LDL) and HDL from PLTP−/− or littermates (N = 3). Data were expressed as the mean ± SD
Discussion
S1P is a key lipid signaling molecule which is closely related to arterial protective functions of HDL [9]. The distribution of S1P in lipoproteins was changed in atherosclerotic diseases [13]. To elucidate the mechanism, we performed this study and found for the first time that: (1) S1P content in plasma was decreased in PLTP−/− mice; (2) PLTP was the key factor to maintain HDL-S1P, and lack of PLTP might decrease HDL-S1P significantly; (3) PLTP could transfer S1P from RBC to HDL, which might be the mechanism by which PLTP affects S1P content in plasma and HDL; 4) the content and distribution of S1P adaptor, apoM, was not affected by PLTP expression.
The distribution alternation of S1P in HDL pool and non-HDL pool might be the important pathogenic factors of atherosclerotic diseases for the reason that S1P can exert quite diverse effects based on where S1P exists in HDL or non-HDL [23]. Thus, the comprehension of factors affecting plasma S1P distribution is critical to the research on atherosclerotic diseases, such as CHD and stroke. HDL is the primary particle of S1P carriers in plasma and the endothelial protective effects via S1P receptors, including barrier function and nitric oxide synthesis, are the important parts of anti-atherosclerotic activity mediated by HDL [9, 24]. Other beneficial effects of HDL-S1P on the cardiovascular system were well documented [3]. As a result, the content and distribution of plasma S1P is critical to HDL functions. Our findings were consist with previous reports that HDL level is the major cause to maintain plasma S1P content [25]. Therefore, enzymes or proteins involving HDL metabolism may change the S1P distribution, thereby participate in the pathogenesis of atherosclerotic diseases.
PLTP belongs to the lipid transfer protein family and transfers amphiphilic molecules among lipoproteins and cells [26]. The level of HDL is dramatically decreased in PLTP−/− mice [10, 18], indicating that the quantity and ratio of proteins as well as lipids might be changed in lipoproteins. PLTP overexpression in apoE−/− mice aggravated atherosclerosis, whereas PLTP deficiency attenuated plaque accumulation in different atherosclerotic models, indicating that the PLTP showed a tight relationship with atherosclerosis [27, 28]. However, no report focuses on the S1P content and its effects on the cardiovascular system from PLTP deficient mice. In this study, we found for the first time that PLTP deficiency decreased the S1P content in both plasma and HDL, suggesting that PLTP is the key factor to preserve HDL-S1P. Moreover, no report focuses on S1P transport from erythrocytes, the major source of plasma S1P, to HDL, the major S1P carrier. Therefore, to further understand the mechanism of plasma S1P transport, an in vitro transfer study is required to identify the role of PLTP on S1P transport.
PLTP can mediate the exchange of several types of lipid molecules, such as phospholipid or vitamin E, in a “lipoprotein–lipoprotein” or “lipoprotein-cell” manner. The relationship between structure and function of PLTP was reported previously [26]. The common property of the “cargos” transferred by PLTP is the amphipathic molecules, with hydrophilic head group followed by a hydrophobic tail. S1P is the phosphorylation metabolite of sphingosine, suggesting that its structure characteristics are similar to other amphipathic molecules transferred by PLTP [14]. Therefore, we performed a S1P transfer assay employing erythrocytes as S1P donor and HDL as S1P acceptor. It is well documented that PLTP activity (the capability of transferring phospholipids among lipoproteins or cells) is closely related to plasma HDL metabolism and its activity accessed by an in vitro donor–acceptor system is well developed and widely applied [12]. Therefore, we used this in vitro system to access the S1P transferring activity of PLTP. This method was repeatable and sensitive for S1P transferring quantitative analysis. In the present study, we found that PLTP could transfer S1P from erythrocytes to HDL. Thus, the alternation of S1P distribution affected by PLTP expression might be elucidated by this transferring mechanism. Nonetheless, further evidence is required to prove the detail of this S1P transport, including an “PLTP-S1P” or “PLTP-X-S1P” interaction investigation and transport efficiency assessment.
A well known decisive factor of HDL preserving S1P is the apoM, an endogenous specific S1P adaptor, which is also enriched in HDL [9]. The results of apoM in plasma and lipoproteins indicated that no apoM content change was found, suggesting that the capability of HDL trapping S1P was not affected. Additionally, the apoM profile in PLTP−/− lipoprotein demonstrated that the PLTP deficiency are quite different from other genetic modified mouse models, such as apoE deficiency or LDL receptor deficiency which were observed apoM shifting from HDL to non-HDL, suggesting that PLTP may transfer “free” S1P to non-HDL independent of apoM [22].
In spite of the transactivation of S1P receptors via connective tissue growth factor -ERK1/2-JNK, LDL also showed a strong S1P trapping activity, and this activity is independent of apoM [25, 29]. Furthermore, our data showed that little apoM was observed in non-HDL, suggesting LDL trapping S1P might be via an unknown mechanism. On the other hand, HDL increased myocardial perfusion under basal conditions, whereas S1P inhibited myocardial perfusion through the S1P3 receptor, demonstrating that the activity of S1P is not consist with HDL bioactivities constantly and the exhibition of S1P activities might depend on the receptor distribution and the “transport form” [30]. Non-HDL S1P increasing in myocardial infarction or stable coronary arterial disease patients indicates that the function of the non-HDL-S1P is worthy of further investigation. Furthermore, in CHD patients, increased PLTP activity was reported, suggesting that PLTP might participate in the metabolism of non-HDL S1P in plasma [31].
An important phenomenon worth noting is that the molar ratio of apoAI and S1P is equal to 50–100 in WT plasma, indicating that over 50 HDL particles process one S1P molecule. However, in PLTP−/− plasma, the apoAI content was decreased by 90 %, S1P content was decreased by 60 %, suggesting that S1P might be partially enriched in PLTP deficiency status [10]. To understand how the metabolism of S1P is affected by PLTP in HDL or other lipoproteins, the content of major apolipoproteins in HDL, such as apoAI, apoAII, and apoM should be considered in further investigation.
In summary, our studies characterized the role of PLTP on the distribution of S1P in lipoproteins and revealed that the mechanism might be due to the transfer activity of PLTP. These findings provided experimental evidence on the pathogenesis research of atherosclerotic cardiovascular diseases.
Abbreviations
- apoM:
-
Apolipoprotein M
- CHD:
-
Coronary heart disease
- HDL:
-
High density lipoprotein
- LC–MS/MS:
-
Liquid chromatography-tandem mass spectrometry
- PLTP:
-
Phospholipid transfer protein
- RBC:
-
Red blood cell
- S1P:
-
Sphingosine-1-phosphate
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TC:
-
Total cholesterol
- TRL:
-
Triacylglycerol rich lipoprotein
- WT:
-
Wild type
References
Hajjar DP, Gotto AM Jr (2013) Biological relevance of inflammation and oxidative stress in the pathogenesis of arterial diseases. Am J Pathol 182:1474–1481
Kontush A, Chapman MJ (2006) Functionally defective high-density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol Rev 58:342–374
Takuwa Y, Okamoto Y, Yoshioka K et al (2012) Sphingosine-1-phosphate signaling in physiology and diseases. BioFactors 38:329–337
Fisher EA, Feig JE, Hewing B et al (2012) High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol 32:2813–2820
Yatomi Y, Ozaki Y, Ohmori T et al (2001) Sphingosine 1-phosphate: synthesis and release. Prostaglandins 64:107–122
Levkau B (2013) Cardiovascular Effects of Sphingosine-1-Phosphate (S1P). Handb Exp Pharmacol 216:147–170
Hanel P, Andreani P, Graler MH (2007) Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J 21:1202–1209
Pappu R, Schwab SR, Cornelissen I et al (2007) Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 316:295–298
Christoffersen C, Obinata H, Kumaraswamy SB et al (2011) Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci USA 108:9613–9618
Jiang XC, Bruce C, Mar J et al (1999) Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J Clin Invest 103:907–914
Schlitt A, Bickel C, Thumma P et al (2003) High plasma phospholipid transfer protein levels as a risk factor for coronary artery disease. Arterioscler Thromb Vasc Biol 23:1857–1862
Attia N, Nakbi A, Smaoui M et al (2007) Increased phospholipid transfer protein activity associated with the impaired cellular cholesterol efflux in type 2 diabetic subjects with coronary artery disease. Tohoku J Exp Med 213:129–137
Sattler KJ, Elbasan S, Keul P et al (2010) Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res Cardiol 105:821–832
Garcia-Pacios M, Collado MI, Busto JV et al (2009) Sphingosine-1-phosphate as an amphipathic metabolite: its properties in aqueous and membrane environments. Biophys J 97:1398–1407
Wang D, Han J, Yu Y et al (2011) Chitosan oligosaccharide decreases very-low-density lipoprotein triglyceride and increases high-density lipoprotein cholesterol in high-fat-diet-fed rats. Exp Biol Med (Maywood) 236:1064–1069
Schmidt H, Schmidt R, Geisslinger G (2006) LC-MS/MS-analysis of sphingosine-1-phosphate and related compounds in plasma samples. Prostaglandins Other Lipid Mediat 81:162–170
Zong C, Yu Y, Song G et al (2012) Chitosan oligosaccharides promote reverse cholesterol transport and expression of scavenger receptor BI and CYP7A1 in mice. Exp Biol Med (Maywood) 237:194–200
Post SM, de Crom R, van Haperen R et al (2003) Increased fecal bile acid excretion in transgenic mice with elevated expression of human phospholipid transfer protein. Arterioscler Thromb Vasc Biol 23:892–897
Argraves KM, Argraves WS (2007) HDL serves as a S1P signaling platform mediating a multitude of cardiovascular effects. J Lipid Res 48:2325–2333
Albers JJ, Vuletic S, Cheung MC (2012) Role of plasma phospholipid transfer protein in lipid and lipoprotein metabolism. Biochim Biophys Acta 1821:345–357
Bode C, Sensken SC, Peest U et al (2010) Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J Cell Biochem 109:1232–1243
Faber K, Axler O, Dahlback B et al (2004) Characterization of apoM in normal and genetically modified mice. J Lipid Res 45:1272–1278
Sattler K, Levkau B (2009) Sphingosine-1-phosphate as a mediator of high-density lipoprotein effects in cardiovascular protection. Cardiovasc Res 82:201–211
Wilkerson BA, Grass GD, Wing SB et al (2012) Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: high density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J Biol Chem 287:44645–44653
Murata N, Sato K, Kon J et al (2000) Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J 352(Pt 3):809–815
Oram JF, Wolfbauer G, Tang C et al (2008) An amphipathic helical region of the N-terminal barrel of phospholipid transfer protein is critical for ABCA1-dependent cholesterol efflux. J Biol Chem 283:11541–11549
Parra ES, Panzoldo NB, Kaplan D et al (2012) The I405 V and Taq1B polymorphisms of the CETP gene differentially affect sub-clinical carotid atherosclerosis. Lipids Health Dis 11:130
Jiang XC, Qin S, Qiao C et al (2001) Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat Med 7:847–852
El-Shewy HM, Sohn M, Wilson P et al (2012) Low-density lipoprotein induced expression of connective tissue growth factor via transactivation of sphingosine 1-phosphate receptors in mesangial cells. Mol Endocrinol 26:833–845
Levkau B, Hermann S, Theilmeier G et al (2004) High-density lipoprotein stimulates myocardial perfusion in vivo. Circulation 110:3355–3359
Robins SJ, Lyass A, Brocia RW et al (2013) Plasma lipid transfer proteins and cardiovascular disease.The Framingham Heart Study. Atherosclerosis 228:230–236
Acknowledgments
This work was supported by Natural Science Foundation of China (81070247, 81170785) and Taishan Scholars Foundation of Shandong Province (zd056, zd057).
Author information
Authors and Affiliations
Corresponding author
Additional information
Y. Yu, S. Guo, Y. Feng have contributed equally to this work.
Electronic supplementary material
11745_2013_3850_MOESM1_ESM.tif
The genotype and phenotype of PLTP−/− mice and their WT littermates a The PLTP knockout was confirmed by agarose gel electrophoresis of PCR products with a single 300 bp band in WT (lane 1), one 240 bp band plus one 300 bp band in PLTP heterozygous (lane 2), and one single 240 bp band in PLTP−/− homozygous (lane 3). b Plasma PLTP activity of WT or PLTP−/− was determined through the transfer of fluorescent phospholipids from donor to acceptor particles reported in reference (12). (TIFF 1 505 kb)
11745_2013_3850_MOESM2_ESM.tif
Ultracentrifugation handling On day 1, 600μl plasma from PLTP−/− or their littermates was loaded into ultracentrifuge tube, and brought to 6 ml with density buffer (d = 1.006 g/ml). The centrifuge tubes were hot-sealed and centrifuged for 24 h at 40,000 rpm at 10 °C. On day 2, the supernatant in fractions 1 and 2 (designated as VLDL1 and VLDL2, respectively) was harvested, and the density of the remnant solution was adjusted to d = 1.063 g/ml and brought to 6 ml. The centrifuge tubes were hot-sealed and centrifuged for 48 h at 40,000 rpm at 10 °C. On day 4, the supernatant in fractions 1 and 2 (designated as LDL1 and LDL2, respectively) was harvested, and the density of the remnant solution was adjusted to d = 1.063 g/ml and brought to 6 ml. The centrifuge tubes were hot-sealed and centrifuged for 48 h at 40,000 rpm at 10 °C. On day 6, the supernatant in fractions 1 and 2 (designated as HDL1 and HDL2, respectively) was harvested. Fractions 3–6 were also harvested and designated as F3, F4, lipoprotein deficiency plasma (LDP)1, and LDP2, respectively. Protein concentration and S1P content of every fraction was determined. (TIFF 423 kb)
About this article
Cite this article
Yu, Y., Guo, S., Feng, Y. et al. Phospholipid Transfer Protein Deficiency Decreases the Content of S1P in HDL via the Loss of its Transfer Capability. Lipids 49, 183–190 (2014). https://doi.org/10.1007/s11745-013-3850-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11745-013-3850-y